您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-06-07 13:17
[摘要]长效注射剂不仅要符合注射剂一般质控要求,还需结合其释放机制和制剂特点进行针对性的质量研究与控制。本文对已上市长效注射剂(化学药品)的药学信息进行总结分析,提出对此类制剂质量控制的一般考虑,为研发者提供相关产品的质控思路。
1概述
长效注射剂( long-acting injection, LAI)通常经皮下或肌内注射等途径给药,在注射位点形成药物储库发挥长效释放作用[1-2]。LAI能够在几天至几个月内持续释放药物,其优点是降低给药频率、简化药物治疗方案、药动学波动较小、不良反应更少。但LAI的应用存在一些局限性,如需就医给药、注射部位疼痛和红斑、可能存在剂量突释、不同制剂间的变异等[3]。正因为存在这些临床或者药学开发方面的挑战,从最早的LAI上市30多年以来,只有20多种LAI化学药品上市,主要用于抗精神病、激素治疗和戒毒等领域。按照释放机制化学药品类LAI大致可分为3类:①原料药缓释,其原料药一般水溶性较差,通过原料药缓慢的溶解和释放发挥缓释作用。②辅料缓释,其释放机制是辅料在给药部位缓慢持续扩散或降解,进而实现原料药长效释放。③药物自凝集缓释,其机制是原料药在体内凝集形成凝胶状药物储库发挥长效释放作用[2]。经初步统计,自1986 年以来共上市26 种化学药品类LAI,其中包括8种肽类药物,基本信息见表1。

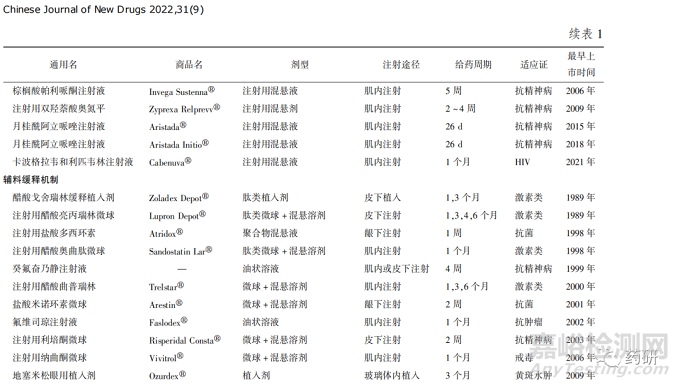
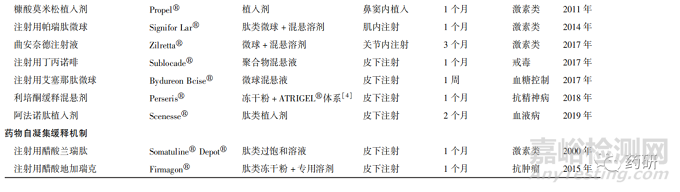
从以上数据中发现,LAI临床适应证的应用相对较窄,加之现有缓释技术及材料发展的局限性,限制了此类制剂的上市数量。 目前LAI产品已开始逐步向抗肿瘤和抗艾滋病等适应证领域扩展,未来产品应用前景可能会更广阔。 由于产品研发难度大、开发周期长,已有LAI均在国外首次上市,国内自主研发经验与能力相对欠缺。 本文主要针对化学药品LAI进行探讨,疫苗佐剂和生物制品不在此处论述。基于国内LAI开发研究基础薄弱的现状,结合自身实际审评工作,提出LAI质量控制的一般考虑,以期为研发和申报提供借鉴。
2质量控制的一般考虑
LAI 的开发应遵循ICH Q8的基本思路,基于拟定适应证的疾病特点和用药需求确定产品目标质量概况(QTPP),进而确定影响产品质量的关键质量属性(CQA),通过原辅包控制、生产工艺控制、中间体控制、终产品质量控制等多种手段,保证产品质量稳定可靠。除需要满足常规注射剂的基本质量要求, 如性状、鉴别、有关物质、装量或者含量均匀度、细菌内毒素和无菌等,LAI的质量控制还应结合其自身释放特点、制剂特性等,进行针对性的质量研究与控制,主要研究内容一般包括下列内容。
ICH Q14最新分析方法开发和Q2分析方法验证
药品研发质量管理体系建立、实施及案例分享
2.1原料药相关质量控制
原料药的基本理化性质决定了此类制剂开发策略的选择,普通制剂通常会选择原料药稳定且溶解度较好的类型。 而原料药缓释机制的LAI 一般开发为注射用混悬液,其往往选择水溶性差但是热力学稳定的原料药,通过混悬液中原料药颗粒在体内缓慢溶解和释放发挥长效作用,因此与原料药溶解度相关的盐型、晶型和粒径分布是控制的关键。
产品开发中应充分研究原料药的成盐类型,加强影响盐型的关键工艺参数控制,确保盐型和比例的一致,并通过多种技术手段确认盐型的准确、稳定。对于含结晶水的原料药,除常规水分检测外,应通过多种检测方法论述结晶水的个数,如单晶衍射、热分析等。晶型方面需关注混悬注射剂中原料药的实际晶型,并进行适当控制。常规采用 X 射线粉末衍射对晶型进行定性分析。此外,通过性状、单晶 X 射线衍射法、热分析、红外光谱、拉曼光谱、显微镜观察等方法也可以进一步分析、确证和控制晶型。晶型稳定性的研究也非常重要,应关注与生产工艺、配伍操作等对晶型稳定及转晶的影响,如研磨、配液、辐照灭菌、冻干等。当原料药存在混晶的情况,应开发混晶定量检测的方法,并研究晶型比例对体内外释放行为的影响。
原料药混悬制剂粒径及粒径分布测定有多种方法,目前主流采用激光粒度仪测定,常见湿法测定。湿法测定中分散介质、分散时间、振摇强度、循环参数等均可能对原料药实际粒子形态及检测结果产生影响,在实际药学研究工作中要注意对粒径检测方法的验证,确保方法准确可靠,重点关注分散样品的稳定性、均一性以及检测方法的精密度和准确度。此外,通过扫描电子显微镜(SEM)和光学显微镜等技术对颗粒形态进行考察,也是粒径研究的重要辅助手段。
对于药物自凝集类LAI,这类产品是由原料药自身交联或者水合在体内形成凝胶储库从而发挥缓释作用,如醋酸兰瑞肽注射液和注射用醋酸地加瑞克。 其与原料药本身的交联特性相关的质量参数均应作为质量控制的重点。 以注射用醋酸地加瑞克为例,要控制肽类原料药醋酸地加瑞克的初级和次级结构、凝集状态;关注配伍药液各关键理化指标的控制,包括药物浓度、性状、复溶时间、光密度、黏度等; 此外,还应进行凝胶动力学和释放度的相关研究[5]。
2.2 辅料相关质量控制
用于注射途径的辅料,在满足药用要求的前提下,通常需要对细菌内毒素和微生物限度进行控制。 辅料缓释的LAI属于典型的通过制剂技术达到缓释效果的制剂类型,主要类型包括微球、植入剂、聚合物溶液和油溶液等,缓释基质通常选择可生物降解的丙交酯乙交酯共聚物(PLGA)、乙交酯共聚物(PLA) 或油类。目前已上市的18种辅料缓释的LAI中,选择PLGA作为缓释辅料的有15种,占比超过80%。
PLGA是一种可生物降解的聚合物,在人体内能够降解成丙交酯和乙交酯分子。根据其聚合程度可分为多种型号。PLGA的聚合物组成( 丙交酯-乙交酯比例)、重均分子量、分子量分布、PLGA结构( 如线性或星形-分支PLGA) 等均可能影响药物体内释放行为[6 - 7], 开发中应评估不同型号( 如7525和5050等)、不同特性PLGA对于微球载药量、粒径分布以及释放度的影响。考虑到稳定性期间PLGA可能会发生水解导致分子量降低,进而影响其他辅料特性,因此还应充分评估其对稳定性末期制剂产品质量的影响,特别关注释放度及其可能引起的体内行为差异。结合临床试验批次数据以及稳定性放置考察结果,合理制定辅料内控标准。为更好地控制产品质量,必要时可以考虑将PLGA的分子量控制纳入制剂质量标准。油类作为缓释辅料的品种较少,以氟维司群注射液为例,蓖麻油是影响氟维司群扩散和吸收的关键辅料,建议严格蓖麻油的内控标准,关注影响释放的关键指标( 如脂肪酸组成、黏度、游离脂肪酸、水分等) 控制。
2.3 释放度
释放度是LAI 常规质控项目。LAI通常采用多点法制定溶出限度,一般包括早期表征突释、表征产品性能稳定以及表征释放完全的时间点。 研发者应当积累临床期间,特别是关键临床批次的溶出曲线数据,采用更多取样点充分表征LAI的体外释放行为。 最终根据多批临床样品的溶出曲线,确定限度标准。 应积累临床批次的生物利用度数据,如果释放行为显著影响生物利用度,设置的释放度限度应能剔除生物利用度不好的批次。
应结合释放机制开发释放度方法,释放度方法应对可能影响体内释放行为的关键理化特性和工艺参数具备一定的区分力,如原料药缓释LAI的原料药粒径分布、形态、晶型以及混晶比例, 辅料缓释LAI的辅料分子量、类型和组成比例,自凝集 LAI的药液浓度等。 如灭菌工艺可能对产品质量产生影响,释放度方法也应对不同灭菌工艺参数具有一定区分力。 鼓励研发者选择更多的物料特性及工艺参数进行全面的筛选研究,以加深对产品性能的了解。
LAI 与常规释放度研究最大的差异在于较长的释放时间。这对于药物本身、释放度试验装置以及释放介质都提出了新的要求:①应充分证明药物在介质中的稳定性能够支持释放度试验的开展,以保证溶出度结果的定量准确。②药典收载的多种释放装置应用于LAI中有其各自的优缺点。桨法和篮法是较为常用的方法,与往复筒法相似,长期释放研究介质挥发的影响可能较大,而且这类装置需要大量的溶出介质。流通池法具有较好的重现性和区分力,但设备昂贵且长期释放研究( 如1 ~ 6个月) 对设备具有挑战。监管机构批准了多种非药典LAI 释放度检测方法, 如透析池法、转瓶法、摇床法等[8]。这些方法属于非标准化的检测方法,设备参数的调整可能会引入较大偏差。例如用摇床法使用不同形状和材质( 吸附性能不同) 的孵育容器、释放介质以及摇晃强度均会对药物释放产生影响,这些由试验方法引入的偏差甚至能够掩盖处方和工艺变化带来的体外释放行为的差异[9]。③ LAI体外释放度检测时间较长,需要确保试验期间释放介质无菌,如注射用艾塞那肽微球的释放试验长达52d,其释放介质中使用的叠氮化钠能够发挥一定的抑菌作用。
非药典释放度方法开发时应关注试验条件考察,包括装置振摇方式、释放容器差异、样品加入和取样方法等。 应考察不同实验室、试验装置略有差异、方法参数适当变化等多种条件对释放度检测的影响,进一步确认方法的适用性及耐用性。 例如,混悬型LAI取样的样品溶液中可能含有药物的不溶性颗粒,需要离心或过滤分离不溶性颗粒,并评估不溶性颗粒返回介质对释放度检测结果的影响[10]。 表2 总结了部分已上市 LAI的释放度检测方法。

通过已上市品种释放度方法的梳理,LAI的释放度最长取样时间一般达到3d以上, 最长可达1个月以上。 与给药周期相比,一般释放度取样时间较给药周期短,但也有部分产品释放度取样时间与药品给药周期基本一致, 如Zyprexa Relprevv®和Zoladex Depot®。还有产品释放度取样时间较给药周期更长,如2017年获批的注射用艾塞那肽微球给药周期为1周,但是释放度取样时间长达52d。取样时间是体外释放度良好控制方法的重要参数。LAI 研发者在释放度检测方法的开发过程中,一般希望能够建立体外⁃体内相关性(IVIVC),以期通过释放度 IVIVC 的建立量化制剂在体内的释放和吸收,并作为有效的质控方法支持在临床开发的后期或上市后处方变更的生物等效性豁免。但由于药物在体内行为的复杂性及体外释放度方法的局限性, 使得体外释放度 IVIVC 的建立具有较大挑战。体外释放度方法作为质控手段,应关注区分力研究,包括关键工艺参数和关键质量属性等[11 - 12]。研究筛选取样时间短、稳定可靠的释放度方法,不仅减少试验时间和成本,也能够提高企业后续生产、放行的效率。
2.4 无菌和细菌内毒素检查
作为注射途径的LAI,无菌及细菌内毒素均为药典明确要求的质控项目。 对于某些特殊类型的LAI,如植入剂和微球制剂由于其所用缓释辅料通常水溶性较差,常规仅能检测该类制剂表面的无菌和细菌内毒素情况,制剂内部则无法检测,因此需关注制剂整体的染菌情况已有研究显示采用溶剂二甲基亚砜可完全溶解微球,球内无菌和细菌内毒素检查方法可行。混悬液的LAI制剂原料药水溶性也较差,也应关注原料药颗粒内部无菌和内毒素检测问题。应注意所选择的检测方法经过了适当的方法学验证,特别关注稀释溶剂对样品检测的干扰。
2.5 其他
混悬型LAI储存过程中会形成松散沉积物,需关注可重悬浮性,对颗粒的分散和凝集状态进行研究。 混悬液还需要考虑配套针头的通针性, 在满足通针性要求的前提下尽可能使用细的针头减轻患者疼痛。 辅料缓释型LAI制备过程中可能使用有机溶剂,制剂质量标准中应设置相应的残留溶剂控制项目。 对于使用专用给药装置的产品,还需要关注给药装置功能性的研究,例如预充针的最大注射力等。 有些植入剂需要特殊的药械给药,药械部分可能需要由医疗器械技术审评中心进行审评。
3小结
LAI作为一类可延长药物作用时间、减少给药次数、改善患者顺应性的制剂,正在成为国内外新药研发的热点。开发者应结合原料药基本理化性质、临床使用特点、目标应用人群等全面分析选择LAI作为目标制剂开发的合理性和可行性。将质量源于设计的理念融入整体开发策略,特别关注LAI释放机制的设计与研究,基于释放机制建立质控前移的思路,根据LAI质控的关键质量属性,评估原辅包和生产工艺对产品质量的影响,并明确控制策略。通过评价历史批次的波动或者试验设计更宽范围的变化,更全面评价关键质量属性和关键工艺参数对产品质量的影响,这些研究工作也有助于更好理解产品特性,支持临床期间以及上市后变更研究。

来源:中国新药杂志


