您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2018-02-07 19:15
一.确定产品范围
医疗器械指令(MDD)覆盖了除体外诊断器械(98/79/EC)和有源植入式医疗器械指令(90/385/EEC)所涉及的医疗器械外的所有器械。
它对医疗器械的定义是指:“制造商的预期用途是为下列一个或多个特定目的用于人类的,不论单独使用或组合使用的仪器、设备、材料或者其他物品,包括所需要的软件。这些目的是:疾病的诊断、预防、监护、治疗或者缓解; 对损伤或者残疾的诊断、治疗、监护、缓解、补偿;对解剖或者生理过程的研究、替代、调节; 妊娠控制。其作用于人体体表或体内的主要设计作用不是用药理学、免疫学或代谢的手段获得,但可能有这些手段参与并起一定辅助作。”对于我国的医疗器械出口制造商来说,明确判断产品的预期用途十分关键。以眼镜架为例,如果用于配制近视镜或远视镜,则属医疗器械;但如果宣称用于太阳镜,则不属于医疗器械。除通过MDD中的定义判定产品外,企业还可借助通用医疗器械命名系统(UMDNS)。该系统建立了7500余种医疗器械的定义和编码,企业可通过检查产品是否存在对应代码来判定其是否属医疗器械范畴。
二.确定产品适用的基本要求
医疗器械指令(MDD)的附录I中规定了所有医疗器械必须满足的基本要求,涉及产品的所有方面。
基本要求共包括14项,其中前6项为通用要求,适用于所有器械,后8项为特殊要求,对某些产品可能部分适用。
(1)通用要求
a.器械必须是安全的,任何风险与器械提供的益处相比必须在可以接受的范围内;
b.器械必须根据最新的知识设计,风险应被消除或预防,最起码要给予警告;
c.器械必须具有制造商规定的性能;
d.器械的安全和性能必须在器械的使用寿命内得以保证;
e.器械的安全和性能在合理的运输、存储条件下必须不受影响;
f.任何的副作用和器械提供的益处相比必须在可接受的范围内。
(2)特殊要求:
a.化学、物理学和生物学特性;
b.传染和微生物污染;
c.结构和环境特性;
d.具有测量功能的器械;
e.辐射防护;
f.对连接或装配能源的医疗器械的要求;
g.制造商提供的信息;
h.临床资料。
三.确定产品适用的协调标准
欧盟委员会授权欧洲标准化委员会(CEN)和欧洲电工标准化委员会(CENELEC)制定了医疗器械指令(MDD)的协调标准,作为满足指令要求的详细技术规范和定量指标。采用协调标准,是制造商使其产品满足指令的基本要求、顺利进入欧盟市场的最为便捷和节约成本的途径。
医疗器械指令(MDD)的协调标准约250余项,其中较为重要的标准包括:EN ISO 13485 质量体系; EN ISO 10993 系列 生物学评价;EN ISO 14971 风险管理;EN ISO 14155系列 临床调查;EN 556系列 灭菌;EN ISO 11607系列 灭菌包装;EN 980 标签符号;EN 1041 医疗器械术语、符号和信息;EN 1174系列 微生物测试;EN 60601系列 医用电气安全。所有医疗器械指令(MDD)的协调标准均由欧盟委员会通过官方公报(OJ)发布,制造商可通过网站获取最新公布的MDD协调标准列表[注1]。对某器械来说,当不存在适用的协调标准时,制造商可按照以下优先极顺序采用标准,作为符合指令基本要求的依据:欧洲标准、ISO/IEC国际标准、欧盟成员国的国家标准、欧盟成员以外的国家标准和企业标准。
四.确定合格评定程序
所有器械通过医疗器械指令(MDD)附录IX的分类规则被划分为四个管理类别:I类,IIa类,IIb类和III类。医疗器械指令(MDD)对每个管理类别内的器械规定了相应的合格评定程序。制造商应该根据附录中的判断规则判断器械属于哪一类别。医疗器械指令对各类器械合格评定程序的要求如以下四图所示,详细规定可参见附录II~VII。
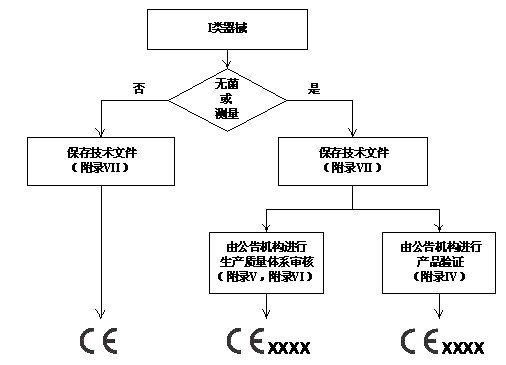
I类设备的合格评定程序
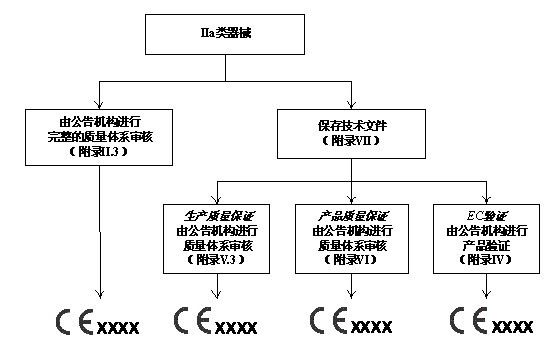
IIa类设备的合格评定程序
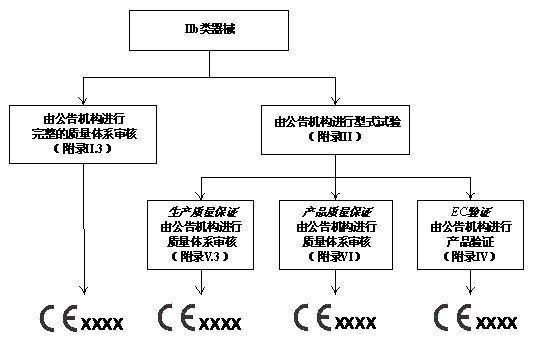
IIb类设备的合格评定程序
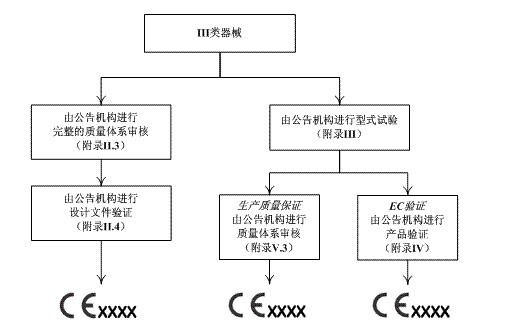
III类设备的合格评定程序
对于低风险的I类器械,其合格评定可由制造商单独完成。对于IIa类、IIb类和III类医疗器械,以及无菌或测量功能的I类器械,MDD要求公告机构强制介入其合格评定过程。此外, 对于IIa类、IIb类和III类器械,MDD提供了两种合格评定模式供制造商选择:通过全面质量体系的设计保证或者通过型式试验的设计保证。
五.准备技术文件
技术文件是证明器械符合医疗器械指令(MDD)基本要求的文件化证据。特别是对I类和IIa类器械来说,由于这两类器械的合格评定过程通常都没有公告机构参与来验证器械的设计,因此技术文件便作为这两类器械的EC合格声明和采用CE标志的制造商的证明。医疗器械指令(MDD)要求制造商必须在最后一件产品制造后将其保存至少5年,以供各成员国主管当局查验。从产品投放欧盟市场起,至少要有一套技术文件必须保存在欧盟境内。医疗器械指令(MDD)的附录II~VII中明确地描述了技术文件应包含哪些文件。技术文件主要包括产品介绍、质量管理体系文件、设计文档、基本要求检查表、风险管理报告、产品包装、标签和随机文件、临床资料、产品型式试验、包装验证报告、灭菌验证报告、如果该产品适合其他设备配合使用,整体符合基本要求的证明材料、是否结合人源性组织或物质的声明、EC符合声明、与欧共体授权代理的协议、CE证书等方面。技术文件应当采用英文或欧盟成员国的官方语言编写。
六.选择公告机构
医疗器械指令(MDD)规定,对IIa、IIb和III类医疗器械,以及无菌或测量功能的I类器械,需要由一个公告机构参与其合格评定程序。目前,被授权依据医疗器械指令(MDD)要求开展合格评定的公告机构共79家,这些机构的信息制造商可通过网站上的公告机构列表获得。
七.起草EC合格声明及注册
依据医疗器械指令(MDD)要求完成器械的合格评定后,制造商应起草并签署EC合格声明,承诺产品符合基本要求和相关协调标准的要求,产品可以合法地投放欧盟市场。此外,根据依据医疗器械指令(MDD)第14条,制造商在将其产品投放欧盟某成员国的市场之前,还必须去该成员国的主管当局(Competent Authorities,CA)注册,提供其在该国营业注册地的地址和有关器械的说明;如果制造商在该成员国内没有注册地址,则应委托欧共体内的授权代理负责产品销售,该授权代理须到成员国的主管当局进行注册。

来源:嘉峪检测网


