ADCs(Antibody-Drug Conjugates)可以简单理解成化疗药、单抗药的结合,将抗体作为药物的靶向“定位器”、毒性分子作为药物的“弹头”,并用合适的连接子组装起来。
相对于以往的化疗药或单抗药,ADC既有单抗药物的特异性,又有小分子肿瘤药的高活性和杀伤力,避免毒性分子对其他正常细胞造成损伤,以靶向性,提高抗体的活性的同时,扩大小分子毒素的治疗窗。
ADC药物由单克隆抗体、细胞毒素、连接子三部分组成,靶标、抗体、linker以及cytotoxic payloads(毒素分子有效荷载)是影响ADC药物的四个要素,决定着药物的疗效。
目前在研ADC药物靶点分布主要集中在HER2,EGFR,CD22,CD19,Trop-2等靶点,从适应症分布来看,实体瘤领域为热门研究领域。
1、ADC药物迭代
根据药物构成和开发技术,通常将ADC药物分为三代,其中:第一代ADC药物多采用可裂解连接子,如Mylotarg使用的腙键,存在提前裂解脱靶的问题,连接不稳定。使用的有效载荷有阿霉素、长春碱、卡奇霉素等,存在毒性高低不适配影响用药安全的问题。
抗体多采用鼠源单抗,会导致免疫反应。代表药物是辉瑞和惠氏联合研制的Mylotarg,用于治疗CD33阳性急性髓性白血病(AML)阳性复发,于2000年上市,由于安全性问题,2010年撤回审批,后修改给药剂量和治疗方案,在2017年重新审批上市。
第二代ADC药物主要由美登素类、奥利司他汀类有效载荷与人源化单抗通过连接子偶联。第二代ADC药物采用不可裂解连接子,优化了药物稳定性问题,但不可裂解连接子无法发挥旁观者效应。在研究更充分的基础上,采用美登素类、奥利司他汀类更有效的有效载荷。开始采用人源单抗,减轻药物免疫反应。
代表药物有:武田和西雅图联合研制的Adcetris和罗氏的Kadcyla,两种药物分别在2015和2013年完成审批,适应症分别为毛细胞白血病和系统型间变性大细胞淋巴瘤以及HER2阳性乳腺癌。采用了传统化学方式的偶联,接头稳定性不佳。
第三代ADC药物在二代药物的基础上,进行了进一步优化,采用更差异化的小分子毒性药物和人源单抗,利用小分子药物与单克隆抗体的位点特异性结合,运用定点偶联技术降低了偶联脱落速度,研发毒性更低、药效更稳定的ADC药物。
在第三代药物中,连接子又改回可裂解连接子,与第一代药物的酸依赖连接子不同,三代药物选择酶依赖连接子,有更好的稳定性。代表药物有阿斯利康和第一三共联合研制的Enhertu、罗氏的Polivy、GSK的Blenrep等,第三代药物的批准时间普遍集中在2018年后,适应症普遍为癌症。三代药物的研究优化仍在不断进行,需要攻克脱靶效应、潜在的耐药性等问题。
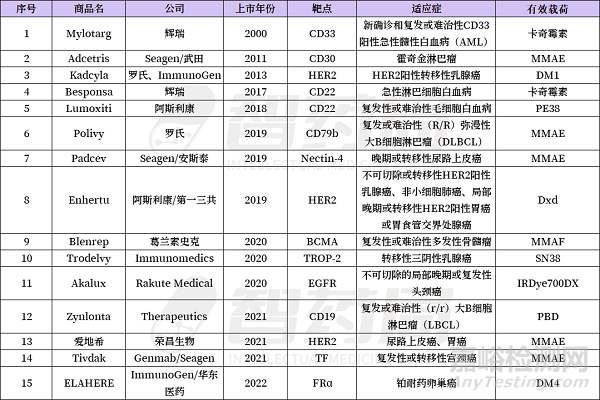
2、15款ADC上市药物
截至2022年底,全球已累计批准上市了15款ADC药物,市场规模已超72亿美元,同比增长31%。其中6款在国内上市,包括罗氏的Kadcyla(恩美曲妥珠单抗)、Seagen/武田的Adcetris(维布妥昔单抗)、辉瑞的Besponsa(奥加伊妥珠单抗)、荣昌生物的爱地希(维迪西妥单抗)、Immunomedics的Trodelvy(戈沙妥珠单抗)、2023年上市的阿斯利康/第一三共的注射用德曲妥珠单抗。
截至2023年一季度,FDA累计批准13款ADC新药。DS-8201今年一季度销量已突破5亿美金,2023年将突破20亿美元成为超级重磅炸弹,超过kadcyla成最畅销ADC也已悬念不大。此外Polivy、Padcev、Trodelvy同样处于快速增长阶段,今年也将大概率超过10亿美元。2023年,全球ADC药物市场规模将大概率超过100亿美元。
在这15款ADC药物中,有6款毒素选择MMAE/MMAF,2款为卡奇霉素类,DS-8021有效荷载为Dxd,PBD类1款。linker方面,采用可裂解linker的有12款,不可裂解linker的3款。
ADC有效荷载
ADC药物大多使用化疗药物作为有效载荷,主要有微管蛋白抑制剂、DNA损伤剂、转录抑制剂等类型。微管蛋白抑制剂可以破坏微管蛋白,影响细胞分裂时纺锤体的形成,干扰细胞分裂;DNA损伤剂作用于细胞DNA,通过破坏DNA结构达到杀伤肿瘤细胞的目的;DNA拓扑异构酶Ӏ抑制剂是目前最具前景的有效荷载,可以裂解单链DNA、抑制拓扑异构酶修复机制,使得DNA损伤,细胞凋亡。除上述三类外还有RNA聚合酶抑制类、凋亡诱导剂等类型的有效载荷。
1、MMAE/MMAF
MMAE(金盏花素E)和MMAF(甲基金盏花素F)是ADC研发最常用的两种奥瑞他汀类化合物具有强效抗肿瘤活性,常用作ADC药物的有效载荷。
Seagen公司针对上述两个毒素分子(微管蛋白抑制剂)进行了一系列专利布局,其中MMAE在美国获得专利保护,期满至2022年11月24日;但在欧洲和中国并没有获得专利保护;MMAF毒素在中国大陆无专利,MMAF+MC在中国2024到期。
MMAE/MMAF的不能用作药物本身,但作为ADC的一部分,连接MMAE/MMAF与单克隆抗体的linker在细胞外液中稳定,ADC与靶向癌细胞抗原结合并进入癌细胞,则被组织蛋白酶切割,之后ADC释放有毒的MMAE/MMAF并激活有效的抗有丝分裂机制。
MMAE/MMAF(金盏花素 Auristatins)在已上市ADC药物中应用最多,其中有5款药物(Adcetris、Tivdak、爱地希、Padcev、Polivy)的有效荷载是MMAE,一款药物(Blenrep)的有效荷载是MMAF。
在2022年的ADC药物销售情况中,SAdcetris营收为14.69亿美元、Padcev营收为7.57亿美元、Polivy营收为4.8亿元,分别排第二位、第五位、第六位。据智药局统计,目前在研的药物以MMAE/MMAF为有效荷载的管线数已超过60个。
2、DM1/DM2/DM4
DM1/DM2/DM4是美登素类微管蛋白抑制剂,可诱导细胞的有丝分裂停止。DM1和DM4常通过使用二硫键与连接子偶联,具有良好的稳定性。
DM1由Immunogen研发,罗氏开发的恩美曲妥珠单抗(T-DM1)使用了该公司技术的特许权使用权,其技术也授权给了安进、礼来、诺华、萨诺菲和武田等企业。2013年罗氏的恩美曲妥珠单抗(T-DM1)在美国上市为标志性事件,意味着ADC药物真正的技术成熟和取得商业化成功。DM1目前在中国专利均已到期。
在上市的ADC药物中,Kadcyla的有效荷载为DM1,2022年营收为22.88亿美元,排ADC药物营收榜第一位;用于治疗铂耐药卵巢癌的全球首创ADC药物ELAHERE™的有效荷载为DM4,于2022年获得FDA加速批准上市。
3、DXd(DNA拓扑异构酶I抑制剂)
拓扑异构酶位于细胞核内,控制和修复在DNA打开、上游转录和复制过程中发生的DNA超螺旋和缠绕。拓扑异构酶I裂解单链DNA,特异性地结合到DNA-拓扑异构酶复合体的界面,抑制拓扑异构酶修复机制,导致DNA损伤,细胞凋亡。
DNA拓扑异构酶I抑制剂(DXd)为喜树碱(CPT)的合成衍生物,最近被用作ADC有效载荷,因为它们具有中等的细胞毒性效力,DXd在血液中的半衰期短,有助于减少毒副作用的产生,具有强大的细胞膜渗透能力,产生旁观者效应,可杀灭临近的肿瘤细胞,且半衰期缩短。
在上市药物中,阿斯利康/第一三共的Enhertu即选择Dxd载荷,2022年营收达13.17亿美元,排ADC药物营收榜第三位。第一三共的DXd平台是近年来较为突出的创新性技术,小分子毒素DXd的专利已经到期,但是MC-GGFG-DXd的组合使用专利(ZL201380053256.2)于2033年到期。
4.SN38(细胞毒素7-ethyl-10-hydroxycamptothecin)
细胞毒素7-ethyl-10-hydroxycamptothecin (SN-38) ,是喜树碱的合成衍生物,由研究人员于 1966 年从中国喜树中分离出来。SN-38 的专利由 Kabushiki Kaisha Yakult Honsha 科学家于 1982 年申请(US 4473692)。
2013 年,Immunomedics 提交了一项专利,描述了多种ADC,包括一种针对TROP2的ADC,其具有与 SN-38 相连的新型linker(US 9028833)。其中一个名为“CL2A”的linker后来被用于 Sacituzumab govitecan。由Immunomedics研发的ADC上市药物Trodelvy®即采用SN38。
5、PBD(吡咯并苯二氮卓)、PE38、IRDye700DX、卡奇霉素
PBD是一类具有抗肿瘤活性的天然产物,属于DNA烷化剂(calicheamicin)。作用在DNA引起细胞凋亡。PBD的结合对DNA造成很小的形变,不容易引起DNA修复,因此PBD毒性很强。Zynlonta是第一款使用PBD类Payload的上市ADC产品,由ADC Therpaeutics公司开发。
PE38部分源自一种细菌分泌的毒素,这种细菌通过阻断细胞制造蛋白质的能力来杀死细胞,抗体片段具有发现并特异性结合肿瘤细胞的能力,可以选择性地将毒素递送至癌细胞。
阿斯利康研发的ADC药物Lumoxiti选择PE38作为有效载荷,该毒素可以抑制蛋白质的合成,并最终触发细胞凋亡。
IRDye700DX(IR700)是光活化化学物质,Rakute Medical的ADC药物Akalux就采用了IR700作为有效载荷,在药物和癌细胞结合后,再向患者照射近红外激光,通过用690 nm的近红外光激发结合抗体的光活化化学物质IRDye700DX(IR700)来破坏癌细胞,是全球首个光免疫疗法。
在上市的ADC药物中,Besponsa、Mylotarg的有效荷载为卡奇霉素。
6.其他有效荷载
微管溶素(Tubulysins):微管溶素可以抑制微管蛋白聚合,使得分裂进程中细胞的细胞骨架解体、凋亡,是良好的微管蛋白抑制剂。
抗有丝分裂EG5抑制剂:抗有丝分裂EG5抑制剂可以抑制纺锤体驱动蛋白参与细胞周期中心体的分离,阻断细胞分裂,具有抗肿瘤效力。
艾日布林(Eribulin):艾日布林是从亚洲海洋海绵中分离的大环软海绵素b的合成类似物,可以抑制微管的伸长,在ADC药物MORAb-202中被用作毒素分子,并已进入临床阶段。
紫杉醇(Paclitaxel):紫杉醇是天然的抗肿瘤药,可以影响微管蛋白聚合,导致有丝分裂停滞、诱导细胞凋亡,在ADC药物领域也有所应用。
杜卡霉素:杜卡霉素是一种细胞毒性物质,在ADC药物的开发中常在酚官能团进行连接子连接,通过其高活性的环丙烷环与DNA的小凹槽结合,并在N3位置烷基化腺嘌呤,达到杀伤肿瘤细胞的目的。
凋亡诱导剂(Bcl-xL抑制剂):Bcl-xL抑制剂能够阻断Bcl-xL上BH3结合域的药物可以触发癌细胞凋亡,它的过度表达是癌细胞获得凋亡抵抗的机制之一。2017年,AbbVie首次以ADC的形式展示了BcL-xL抑制剂的有效载荷,其靶向表达EGFR的特定细胞或组织。氨基烷基延伸的核心修饰用于在需要时建立合适的连接位点。
RNA聚合酶抑制类(鹅膏蕈碱):amatoxin衍生物具有一个八个L-构型氨基酸组成的大环,通过亚砜部分连接在色氨酸和半胱氨酸残基之间。amatoxins的三个侧链是羟基化的,OH基团具有良好的水溶性并与目标分子结合。两种肽,α-鹅膏糖蛋白和β-鹅膏毒素,占所有毒素的90%。在ADC技术领域,使用类似amatoxins的转录抑制剂是一种相对较新的方法。
泰兰司他丁及其类似物:泰兰司他丁及其类似物可以与剪接体的SF3b亚单位结合,从而阻止RNA剪接。靶向剪接体是一种参与mRNA加工的大型核糖核蛋白复合物,为靶向癌症治疗提供了一种有希望的治疗选择。有几种天然产物能够通过与不同的剪接体亚单位结合来抑制RNA剪接。
烟酰胺磷酸核糖基转移酶:烟酰胺磷酸核糖基转移酶(NAMPT)是一种负责将烟酰胺转化为烟酰胺单核苷酸的酶,其抑制剂在各种临床前和临床研究中显示出有效性,但其临床应用受到靶向毒性和剂量限制性毒性的限制,如血小板减少和胃肠道不良反应。
卡马霉素:从库拉索菌中分离得到两种新的蛋白酶抑制剂,卡马霉素A和卡马霉素B。他们被发现能抑制酿酒酵母20S蛋白酶β5亚单位活性(糜蛋白酶样活性)。此外,它们对肺癌和结肠癌细胞株具有很强的细胞毒性。
但是由于它们的高效价,选择性较差,经常表现出毒副作用。因此,剧毒的卡马霉素衍生物适合作为ADC的弹头,可以保持所需的效力,并获得更好的耐受性。
3、连接子
ADC药物的连接子根据不同的水解方式,分为可切割连接子和不可切割连接子。可切割连接子可以通过细胞中的生理条件释放细胞毒性物质,如二硫化物等;不可切割的连接子只能通过细胞内的溶酶体降解,如无裂解机制的稳定接头。不可切割连接子ADC药物在血液中具有更长的半衰期和更低的脱靶毒性。
第一代ADC上市药物采用可裂解连接子,如Mylotarg 使用了含酸敏感腙键和GSH敏感二硫键的可裂连接子,该连接子在血液循环中可能发生断裂,提早释放产生脱靶毒性,增加了毒副作用。
第二代ADC采用不可裂解连接子,主要加入了马来酰亚胺(MC)结构,降低了脱靶毒性。如Kadcyla 中所使用的马来酰亚胺(MC)连接子,Besponsa中所使用的SMCC连接子等。但第二代ADC由于偶联缺乏特异性,并没有解决药物均一性差,存在未结合抗体等问题,且不可裂解连接子无法发挥旁观者效应,限制了第二代ADC的疗效。
第三代ADC药物的一个里程碑式进展是采用了定点偶联技术,比如2019年获批的Enhertu,Padcev。2019年获批上市的Padcev和Polivy使用了与Adcetris 相同的缬氨酸-瓜氨酸二肽连接子(Val–Cit)。在Enhertu中,基于甘氨酸-甘氨酸-苯丙氨酸-甘氨酸的四肽连接子(MC-GGFG-AM)首次得到应用。研究结果表明,Enhertu具有优异的血液循环稳定性,血液清除率非常低。
4、药物偶联方式
ADC的药物偶联即是指将ADC各组分连接起来的连接方式,决定药物抗体偶联比(Drug Antibody Ratio,DAR)和均一性,影响药物的活性、耐受性和稳定性。一般分为随机偶联和定点偶联两类。
随机偶联:是指是指将细胞毒素或化疗药物随机偶联到抗体分子。不改变抗体,直接利用其表面的赖氨酸残基烷基化或酰化作用,或通过还原二硫键释放半胱氨酸残基与连接子相连,偶联位置和载药数目不确定,产生具有不同DAR的异质混合物,其动力学性质不一、稳定性差,会导致药物毒性增强、治疗窗变窄。
定点偶联:是指将细胞毒素或化疗药物偶联到抗体分子中特定的位置。通过改变抗体实现在抗体特定位点连接载药,提高了药物的均一性大为,降低了毒副作用,拓宽了治疗窗口,还能运用于核素等其他偶联药物的开发,成为 ADC 开发的重要发展方向。
随机偶联和定点偶联的不同效果主要体现在药物抗体偶联比(Drug Antibody Ratio,DAR)。DAR可能在0-8之间变化,理想的DAR为2-4。DAR体现偶联细胞毒素的多少,会影响药物的有效性和均一性。
赖氨酸酰胺偶联:赖氨酸酰胺偶联是一种主要的ADC偶联方法,利用抗体的赖氨酸残基,氨基酸亲核NH2基团与利克有效载荷上亲电的N-羟基琥珀酰亚胺基团发生反应,使用含有活化羧酸酯的连接剂将抗体上的有效载荷和赖氨酸残基连接起来。
半胱氨酸偶联:半胱氨酸偶联是基于半胱氨酸的偶联方法。通过抗体的半胱氨酸残基与在负载上的巯基反应官能团之间反应进行连接。
基因工程掺入非天然氨基酸。基因工程掺入非天然氨基酸是基因工程用反应手柄安装非天然氨基酸残基进行位点特异性反应,引入羰基与烷氧基胺官能化接头反应以发生特异性偶联,用于精确控制ADC药物的DAR。
糖工程特异性偶联:糖工程特异性偶联是指通过将药物连接子与位于CH2结构域的N297糖链偶联,达成ADC的位点特异性偶联。根据糖链的非还原末端存在的不同单糖,像半乳糖、N-乙酰半乳糖胺(GalNAc)、N-乙酰氨基葡萄糖(GlcNAc)等,可以特异性地将ADC连接子连接到这些糖上。