您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-12-19 08:24
前言:这个主题,是关于药物晶体中微量无定型的定量分析,微量无定型定量特指无定型含量<10%情况下的定量分析。药物为什么要做晶型中微量(<10%)无定型的定量呢?因为制剂处理过程(如微粉化、粉碎、冻干、喷雾干燥、研磨和压片),会导致晶体中产生不同程度的无定型,并且产生的无定型主要吸附在晶体表面,影响晶体的表面性能,从而会严重影响产品的后处理和药物的稳定性。目前行业内,做晶型中微量无定型定量的品种,主要集中在吸入粉雾剂品种如“茚达特罗”。
1、 分析检测手段
分析方法的开发,主要考察的是灵敏性、专属性、检测限和定量限。不同检测手段的定量限LOQ和检测限LOD及特点汇总列于表1。目前来说,晶体中微量无定型的检测,行业内检测精度较高且公认的方法有:恒温量热仪isothermal microcalorimetry (IMC); solution calorimetry (SC),固态核磁(ssNMR)和动态水分吸附仪dynamic vapor sorption (DVS)。对于药企来说,这四种检测手段中,最可及的检测手段是动态水分吸热仪(DVS),因此本文主要介绍如何利用DVS做无定型定量方法开发。
表1 无定型定量的分析检测手段及精度汇总表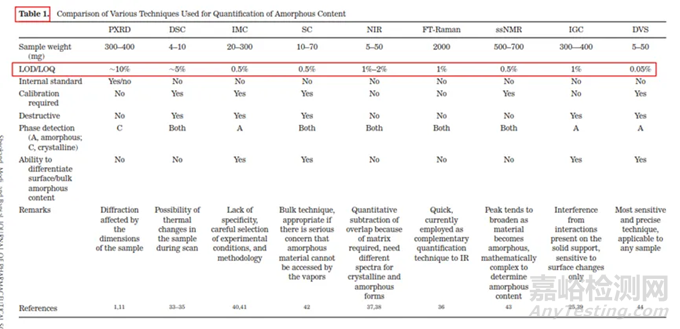
2、 DVS定量分析的三种方法
DVS用于无定型定量的分析,主要有三种方法,概述如下:
方法一(Method 1):平衡吸湿法(Equilibrium Moisture Uptake Method)
原理:测定晶型、无定型、二者混合物的平衡吸湿增重的量;在较宽的无定型比例范围内,平衡吸湿增重与无定型含量呈线性关系。
方法适用性:适用于满足以下条件的情形—a,无定型在重结晶前,样品有较大量吸收增重;b,无定型在选择的实验条件下不发生转晶(如下图1所示)。
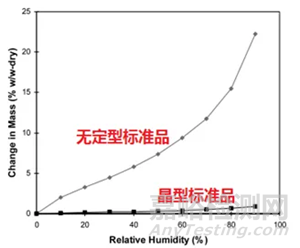
图1 适合选择方法一的典型吸附曲线示意图
平衡吸湿法的检测限受限于选定条件下样品的吸湿增重的量,换句话说,质量变化越大,选择平衡吸湿法做定量分析的准确度越高。
方法二(Method 2):吸收法(Water-Solvent Uptake Method)
原理:恒定湿度下,比较无定型转晶前后的重量变化。前后吸湿重量差和无定型含量呈现线性关系。
方法适用性:在特定溶剂氛围湿度下无定形能重结晶形成晶体的情形。
方法二开发的流程相对复杂一些,但精度更高,具体如下:
(1)测定纯无定型样品吸附曲线(moisture sorption profile)
下图2是无定型转晶的典型的吸附曲线,RH≥60时,无定型发生转晶变成晶体,吸附曲线出现转折。这里需要强调的是,DVS不单单是只能做水分吸附,也可以开发有机溶剂吸附的方法。根据Gordon-Taylor(G-T)公式,水分或者惰性溶剂都是良好的增塑剂(如水的玻璃化转变温度136K,乙醇和丙酮大约为100K),无定型吸收水分或者溶剂后,玻璃化转变温度降低,从而发生转晶。总而言之,开发方法的第一步,是需要寻找合适的测试温度和诱导溶剂氛围,使得纯无定型在选定条件下发生转晶。
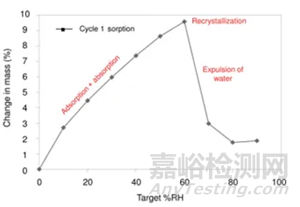
图2 无定型湿度介导转晶示意图
(2)临界湿度点的确定(Critical Point)
临界湿度点定义:在选定条件下,无定型发生转晶的湿度点。上图1中临界湿度点为60%。在临界湿度点前,无定型既有表面吸附(adsorption)也有主体吸收(absorption),无定型转换为晶体后,晶体只有表面吸附,这也是图1曲线出现引湿增重转折点的根本原因。图2给出了表面吸附和主体吸收的示意图。
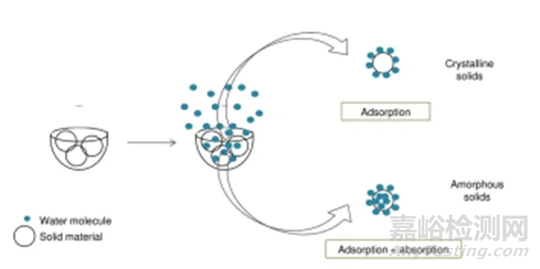
图2 表面吸附和主体吸收的示意图
(3)参考湿度点确定(Reference point)
Reference point是指选定用于测试的湿度点。它需要选择在无定型转晶前,并尽可能选择吸湿增重较大的湿度点。如文献2中,转晶湿度点RH=60%,选择参考湿度点RH=45%。
(4)样品测试及标准曲线绘制
配置不同比例的混合物(无定型+晶型)样品,按类似图3所示方法测试DVS引湿增重:
Step I:RH-=0% 干燥至恒重;
Step II:RH=45%(reference point )足够时间得到吸附吸收平衡;
Step III:RH-=0% 干燥至恒重;
Step IV:RH>临界湿度 足够时间无定型发生完全转晶;
Step V:RH-=0% 干燥至恒重;
Step VI:RH=45%(reference point )足够时间得到吸附吸收平衡;
以Step II和Step VI重量差为纵坐标,无定型含量为横坐标,做图即可的如图4所示是标准曲线。
Method 2的优点:精确度较高,因为直接测定无定型的参数,但如果开发的方法不合适,无定型转晶不完全,则可能导致该方法的定量精度大大折扣。
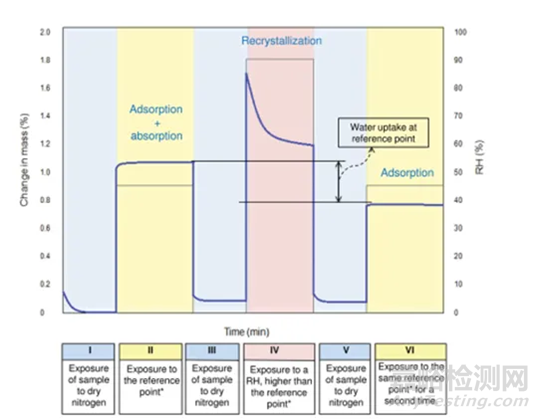
图3 Method 2测试方法示意图
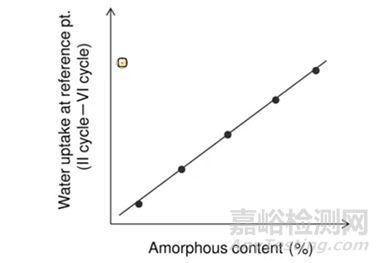
图4 Method 2 标准曲线绘制示意图
方法三(Method 3):剩余重量法(Residual Weight Method)
原理:低湿度下的吸附脱附差值(如图4红色圈所示)与无定型含量呈线性关系。无定型转晶前,在DVS条件下有表面吸附和主体吸收,当转晶形成计量化学比的水合物或者溶剂化物晶体后,进行脱附过程,进入晶格内部的计量化学比的溶剂(或水)不会脱除,那低湿度下的吸附脱附差值仅仅与无定型含量相关,因此可以用来做定量。
方法适用性:无定型在选定的测试条件下,必须能形成计量化学比的化合物或者水合物,所以应用相对受限。
方法3的开发流程与方法2类似。这里不做赘述。该方法精度同样较高,但需要保证无定型在特定湿度下下完全转晶为计量化学比的溶剂化物。
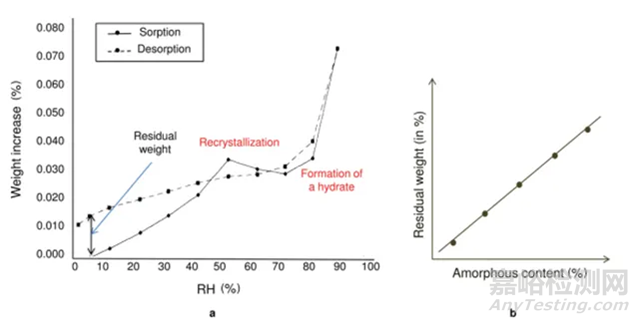
图5 Method 3 测试方法示意图
3、DVS定量分析方法选择的决策树
上面介绍了DVS进行无定型定量的三种方法,其中Method 2和3精度更高,但每种方法均有各自的适用性。下图给出了具体选择哪种方法的决策树。
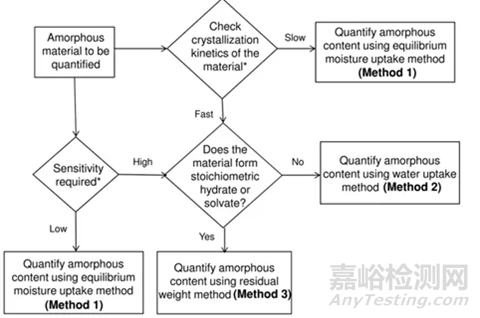
图6 DVS无定型定量方法选择决策树
4、 DVS做无定型定量的注意点
据文献报道,DVS分析方法对于1%以下含量的样品精度也较高,检测限最低能做到0.05%。那DVS分析方法开发中,有哪些注意点呢?
(1)温度
温度越高,吸湿会增加(反例很少),低含量的无定型,吸湿很低,因此需要评估吸附受温度影响的大小;此外对于Method 2和3,都需要无定型能发生转晶过程,对于转晶来说,温度控制也是关键参数。总之,需要对DVS测试的温度进行选择。
(2) 平衡时间:(Equilibration Time)
平衡时间太短,吸附可能未达到平衡,也可能导致转晶不彻底;平衡时间太长,浪费设备资源。因此平衡时间也需要优化方法。下图7是平衡时间对吸附影响的一个案例,可以通过吸附-脱附间隙定性判断平衡时间是否合适。吸脱附间隙不单受平衡时间的影响、还和药物粉末的多孔性、无定型含量和是否形成计量比的溶剂化物有关,需要根据具体情况做好甄别。
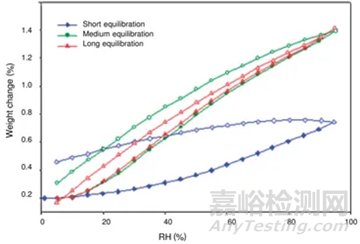
图7 平衡时间对吸脱附“间隙”的影响
(3) 定量用标准品的预处理
100%晶型标准品理想情况下,需要与粉碎后API具有相同的理化性质(这里主要指粒度、形貌和表面特性)。所以一般采用粉碎后(微粉化、球磨等)晶体样品进行预处理后作为100%晶型标准品。预处理操作是:将微粉化样品放置在特定溶剂氛围下(如90%丙酮)使无定型样品充分转晶形或者消除表面晶体缺陷。
100%无定型一般也做对应粉碎处理,然后用XRD/DSC/红外/拉曼等表征,一般需要现配现用。对于DSC测试玻璃化转变温度较高的无定型(稳定性较好的无定型),一般放在P2O5干燥器,尽量低温保存。无定型标准品测试前,需要预干燥,一般放置25℃/RH=0%的干燥器中。
(4)装样量及样品粒度
DVS定量测试,装样量需固定。理论上,引湿增重>10%,5-10mg的装样量做定量就够;引湿增重<0.5% 推荐更大量的装样量50mg±5mg。装样样品要求尽量平铺,最大化的暴露比表面积。
样品的粒度,更倾向于用小粒度做定量,且无定型和晶体有相似的几何参数(粒度和比表面积),形貌和粒度对于Method 1的影响最大,Method 2和Method 3方法的敏感度相对会小一点。
小结:DVS无定型定量,开发最优的参数条件,无定型定量的检测限和定量限可以做到很低。另外,DVS测试不需要样品均匀混合(直接计算标准品比例对应称量质量即可),用样量低,笔者推测行业内会越来越多的使用。
参考文献
[1] Sheokand S , Modi S R , Bansal A K .Dynamic Vapor Sorption as a Tool for Characterization and Quantification of Amorphous Content in Predominantly Crystalline Materials[J].Journal of Pharmaceutical ences, 2014, 103(11):3364-3376.DOI:10.1002/jps.24160.
[2] Shah B , Kakumanu V K , Bansal A K .Analytical techniques for quantification of amorphous/crystalline phases in pharmaceutical solids[J].Journal of Pharmaceutical Sciences, 2006, 95(8):1641–1665.DOI:10.1002/jps.20644.
[3] Mackin L , Zanon R , Park J M ,et al.Quantification of low levels (<10%) of amorphous content in micronised active batches using dynamic vapour sorption and isothermal microcalorimetry[J].International Journal of Pharmaceutics, 2002, 231(2):227-236.DOI:10.1016/S0378-5173(01)00881-X.
[4] Giron D , Remy P , Vilette S T .Quantitation of amorphicity by microcalorimetry[J].Journal of Thermal Analysis&Calorimetry, 1997.DOI:10.1007/BF01979493.

来源:Internet


