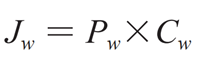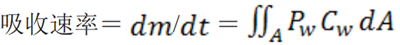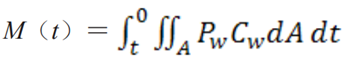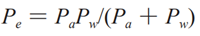人体生物等效性试验豁免(以下简称为“生物豁免”) 是指申请人向药品监管机构提供所申请注册的药品或已上市药品的上市后变更,不适宜进行人体生物等效性试验或者生物等效体外替代试验的相关证明性文件和数据, 从而免予证明该药品在体内的生物利用度和生物等效的可能性。
2017 年 11 月,原国家食品药品监督管理总局仿制药质量与疗效一致性评价办公室经组织人员对 289 种基本药物目录内的品种进行梳理和调研,并报仿制药质量和疗效一致性评价专家委员会审议通过,拟定了首批拟推荐或简化生物等效性试验的品种名单并向社会公开征求意见。
该名单中共包括可采用药学方法评价一致性、 推荐生物豁免的品种 20 种;
可采用药学方法评价一致性, 但企业须自证为生物药剂学分类系统(biopharmaceutics classification system,BCS)分类 1 或 3 类药物,并根据《人 体生物等效性试验豁免指导原则》提交溶解度、渗透性、 溶出度等相关研究资料的品种 20 种 ;
可采用药学方法评价一致性、以药代动力学比较方法评价安全性的品种 4 种;
可仅进行餐后生物等效性研究的品种 1 种;
可仅进行空腹生物等效性研究的品种 12 种。
此为各国药品监管机构中首次公开发布拟生物豁免品种名单。
基于科学的生物豁免可减少不必要的健康人群对药物的暴露、节约一致性评价或仿制药研发的研究成本和时间。
生物豁免在制药行业中的呼声甚高,但对 其科学性的系统理论研究却不多。本文从生物豁免的理论出发,概要阐述可能造成生物不等效的风险因素以及生物豁免的风险评估与管理,供制药企业和监管机构在进行生物豁免决策时参考。
一、生物豁免的适用情况
1 胃肠道局部作用药物
对作用机制明确、不良反应较明确、体内吸收甚低或不吸收、不适合进行以药代动力学参数为终点评价指标的人体生物等效性试验的胃肠道局部作用药物,可申请生物豁免。
如:保护胃黏膜、促进胃黏膜修复作用的铋剂;吸附消化道内病毒、病菌及它们产生的毒素,缓解腹泻的蒙脱石散 ;增加大肠内粪便含水量、缓解成人和 8 岁以上儿童便秘症状的聚乙二醇 4000 散;中和胃酸的碳酸氢钠片等。
2 溶液制剂
外用溶液剂、口服溶液剂、气雾剂、喷雾剂、滴鼻剂等溶液制剂,药品的活性成分呈完全释出状态,当供试品与参比制剂具有相同的处方,且处方中不含任何对药品的活性成分吸收有影响的辅料时,可申请生物豁免。
如果仿制药采用了不同的辅料且可能会影响药品的生物等效时,则不批准其生物豁免申请。
比如,山梨(糖)醇、 甘露醇和木糖醇均为药品中常用的辅料,这些辅料在胃肠道中的吸收较差,会增加肠道的渗透压,影响胃肠道中水分的输送和肠道的运药时间,还可能影响药物跨肠壁吸收的速率,从而影响药物的吸收 ;
具有渗透影响作用的成分对药品在上肠道的停留时间影响较小,而对在下肠道的停留时间影响显著。
3 同一药品的不同剂量规格
对处方相同、活性成分与辅料比例相似且在不同 pH 溶出介质中的体外溶出曲线相似的不同剂量规格的同种药品,通常高剂量规格已做过生物等效性试验的,低剂量规格可申请生物豁免。
有些品种由于安全性或其他原因,可选择较低剂量规格进行体内生物等效性试验。
4 经长期临床有效性验证或具有细胞毒性的药品
对某些药品,如果已经过长期的临床有效性验证或具有细胞毒性,没有必要进行生物等效性试验或因可能对健康人体产生不可逆的损伤,经风险收益比分析,对可通过与参比制剂比较体外溶出曲线的方法来替代生物等效性试验、从而考察仿制药与参比制剂质量和疗效一 致性的,可申请生物豁免。
如美国 FDA 启动的“药物有效性执行方案”(Drug Efficacy Study Implementation)中 的某些品种,包括异烟肼片、泼尼松片、制霉素混悬液等。
5 基于 BCS 分类的豁免
BCS 是基于药物本身的溶解性和渗透性进行药物分类的一种科学系统,由 Amidon 等 于 1995 年提出,用于分析普通口服制剂采用体外试验得出体内生物等效的可行性。
基于 BCS 分类的生物豁免理念自推出以来,已陆续得到美国、WHO 和欧盟等药品监管机构的认可, 并陆续出台了相应的法律法规、指导原则和豁免条件标准 。
我国原国家食品药品监督管理总局也于 2016 年 5 月发布了我国基于 BCS 分类的《人体生物等效性试验豁免指导原则》。
二、BCS
Amidon 等认为,当涉及到口服固体常释制剂中的活性药物成分在体内的吸收速率和程度时,BCS 主要考虑以下 3 个关键因素,即药物溶解性(solubility)、肠道渗透性(intestinal permeability)和制剂溶出度(dissolution)。
根据菲克第一定律,在单位时间内通过垂直于扩散方向的单位截面积的扩散物质流量(扩散通量,diffusion flux,用 J 表示)与该截面积处的浓度梯度(concentration gradient,用 C 表示)成正比,也就是说 C 越大、J 越大。
将此定律用于药物在体内的吸收即可知,药物通过胃肠膜表面的浓度越大,其在胃肠道被吸收的就越多。
公式(1)
式中 Jw(x, y, z, t)为在任何时间和位点通过胃肠壁的药物通量(质量 / 面积 / 时间),Pw(x, y, z, t)为胃肠膜渗透系数,Cw(x, y, z, t)为胃肠膜表面的药物浓度。
公式(1)是以药物在复杂的胃肠道处符合漏槽条件(即溶出介质体积大于药物溶解所需体积的 3 倍以上)和 Pw 是有效渗透系数为前提的。
任何时间点的药物吸收速率即药物从胃肠道减少的速率为:
公式(2)
吸收过程发生于整个胃肠道表面。
在 t 时间点的总吸收质量 M 为:
公式(3)
Pw 随时间不同而不同、吸收位点不同而不同,如十二指肠、空肠、回肠和结肠的表面形态具有随意性, 又如介质环境不同、药物在不同肠壁的转运方式不同等,这些都可能影响药物的渗透性。
基于以上公式,生物利用度可叙述为:如果 2 种不同的药品含有相同的活性成分且在胃肠壁表面具有相同的浓度 - 时间曲线,则它们具有相同的吸收速率和程度。
生物利用度还可进一步叙述为:如果 2 种不同的药品在所有的胃肠腔条件下均具有相同的溶出曲线,则它们具有相同的吸收速率和程度。
这些叙述是以药品处方中不含有可能会影响药物渗透性和(或)肠道转运的其他成 分为前提的。
药品的体内溶出与其在胃肠壁浓度的关系比较复杂,受胃肠道复杂的水动力学和内容物的影响。
公式(4)
式中 Pe 为有效渗透系数,Pw 为此上讨论的胃肠壁渗透系数,Pa 为表观渗透系数。
根据 BCS,药物被分为以下 4 类:
1 类:高溶解性、高渗透性的药物;
2 类:低溶解性、高渗透性的药物;
3 类:高溶解性、低渗透性的药物;
4 类:低溶解性、低渗透性的药物。
三、生物豁免的风险分析与管理
生物豁免可减少人体试验、缩短研发周期、减少研发投入,但生物豁免后的生物不等效风险则可能对药品的安全性或疗效产生不良影响。
在制药企业制定研发策略时,在监管机构审评审批生物豁免申请时,宜综合分析生物等效性的影响因素,对(拟)申请生物豁免的品种进行定量或定性的风险评估,对潜在风险进行预判和应对,从而对是否申请或批准生物豁免进行科学的决策,以减少生物豁免风险事件的发生或减少风险事件带来的不利影响。
1 生物不等效风险分析
1)生物不等效风险低的情况包括:
(1)BCS 分类 1 类药物。国际药学联合会出版的生物豁免专刊中提及,BCS 分类 1 类的药物中有多种药物的口服常释制剂可以予以生物豁免,如阿司匹林、盐酸 多西环素、盐酸普萘洛尔、硫酸伯氨喹、司他夫定、泼尼松、泼尼松龙、左氧氟沙星、甲硝唑、盐酸阿米替林、 盐酸维拉帕米和氯喹。
但硫酸奎尼丁和硫酸奎宁由于治疗指数窄,它们的口服常释制剂不能予于生物豁免。
(2)部分 BCS 分类 3 类药物。根据国际药学联合会出版的生物豁免专刊,BCS 分类 3 类药物中口服常释制剂可以予于生物豁免的药物包括对乙酰氨基酚、阿昔洛韦、阿替洛尔、西咪替丁、乙胺丁醇、异烟肼、拉米夫定、 甲氧氯普胺、吡嗪酰胺和雷尼替丁。
以盐酸二甲双胍片为例。文献记载,盐酸二甲双胍片如在 1 h 内溶出达到 100%,则其可与口服盐酸二甲双胍溶液达到相同的生物利用度。
研究显示,盐酸二甲双胍片是生物不等效风险低的品种,在单点溶出度符合要求的情况下,即使仿制药与原研药的体外溶出曲线差别较大,人体生物等效性试验结果也具有生物等效性。
该例子表明,体外溶出曲线存在差异并不意味着在体内一定生物不等效。
2)生物不等效风险高的情况包括:
(1)大部分 BCS 分类 2 类药物。根据国际药学联合会出版的生物豁免专刊,仅有 3 种 BCS 分类 2 类药物的口服常释制剂可以予于生物豁免,这 3 种药物均为非甾体抗炎药,即双氯芬酸钠 / 钾、布洛芬和酮洛芬,它们均为弱酸性药物,在酸性条件下溶解度低,但在 pH 6.8 的小肠环境中溶解度高。
一项对 500 项生物等效性试验的荟萃分析表明,BCS 分类 2 类药物出现生物不等效的 风险较分类 1 和 3 类药物高 4 倍。
(2)BCS 分类 4 类药物。此类药物由于溶解性和渗透性均低,不能予于生物豁免,属生物不等效风险高的药物。
(3)口服缓 / 控释和定位释放制剂。与普通口服常释制剂相比,口服缓 / 控释和定位释放制剂的处方、工艺更复杂,影响药物溶出或释放的因素更多,进入人体消化道后在不同部位的溶出或释放特点不同,在体内的吸收过程也更复杂,一般也属生物不等效风险高的药品。
对于此类药品,应研究和寻找与体内吸收行为有相关性的体外溶出或释放的试验方法,只有采用具有体内体外相关性的溶出或释放评价方法才能预测其体内吸收行为。
仿制药与参比制剂的多条溶出曲线接近有利于、但不能保证它们体内的吸收速率和程度相似,关键是要有与体内吸收行为具有相关性的体外溶出或释放试验的方法及结果。
(4)其他高风险因素。如治疗窗狭窄且临床使用中不监测血药浓度的药品;用于特殊人群如老人、儿童、孕妇的药品,或属高危、急救药品等;处方中辅料影响吸收的药品 ;质量标准不能良好地反映药品质量的药品;高变异性药品;药代动力学性质呈非线性的药品等。
2 生物豁免的风险分析
从广义上来说,生物豁免风险主要包括:
(1)药学等效、但生物不等效,由此对药品的疗效与安全性产生不良后果。
(2)不同国家的药品监管机构对生物豁免的接受程度不同。比如,目前美国和欧盟已接受基于 BCS 分类的生物豁免,但日本尚无批准先例。
(3)生物豁免常需提前申请,可能会在一定程度上延长药品的申报注册时间。
此外,虽然目前多个国家或组织的药品监管机构已发布了生物豁免相关的指导原则,但还均尚属纲领性指导原则,可操作性不强,申报单位在进行相关研究及申报注册时可能面临不知从何入手的问题,不同的审评审批人员在审评审批时也可能具有不同的见解与判断标准,从而作出不同的审批结论。
3 生物豁免的风险管理
风险管理是指识别出风险点之后把风险可能造成的不良影响减至最低的管理过程。针对以上生物豁免的风险,申报单位或监管机构可采取以下风险管理措施:
(1)如前分析,可能造成生物不等效的影响因素众多,包括药品的药学性质、制剂性质、药代动力学性质等, 且不同影响因素对生物不等效的风险及对药品的安全性和疗效的影响程度也不同。应用科学的定性和(或)定量方法对生物豁免后生物不等效的风险进行评估可为选择进行生物等效性试验还是申请生物豁免提供重要的决策依据。
另外,采用通过监测治疗窗狭窄药品用药后的血药浓度、处方上与原研药保持一致、不采用可能影响药物吸收的辅料等方法也可降低药品的生物不等效风险。
(2)综合分析不同药品监管机构的药品审评审批理念与标准,制定综合性的药品研发与注册策略。
(3)对拟申请生物豁免的品种,宜综合分析生物豁免的可行性,充分准备生物豁免相关的研究资料,并尽量在研发过程中提前与审评审批机构就研究方法等技术问题进行沟通,沟通方法参见原国家食品药品监督管理总局发布的《药物研发与技术审评沟通交流管理办法(试 行)》。
结语
随着人们对口服制剂的性质和药物体内吸收的动力学性质的了解越来越多,加之已有更科学、合理的体外试验方法用于预测药物的体内吸收过程,生物豁免的界定范围将会得到科学的扩大,从而使更多的药品获准生物豁免。
这不仅能为众多的制药公司节约大量资金投入,也能减少健康人体对药物的暴露、缩短仿制药的研发周期及费用,最终对广大患者也是有利的。
文章来源:《上海医药》2018年第39卷第3期
原标题:《仿制药一致性评价中人体生物等效性试验豁免的风险分析与管理》
作者:廖萍 张景辰 李帅 龚前飞 陈桂良