多肽类药物安全性评价是缺少特定指导原则的。ICH M3(R2)更多是针对小分子,ICH S6(R1)则适用于大分子。多肽药物分子量介于两者之间,制备方式既有化学合成,也有发酵所得。选择1998-2019年间FDA批准的46个多肽药物进行分析,其中21个是化学合成的、6个半合成、18个重组表达、1个是天然产品。其中,5个BLA上市,41个NDA上市。本文就多肽类药物一般毒理研究、遗传毒性研究、免疫原性测定、生殖和发育毒性、杂质、安全药理测定、非蛋白原性氨基酸等相关内容进行简单总结分享。
多肽药物从20世纪初就开始用于临床,早期主要是从天然产物中提取,包括激素替代物胰岛素、促肾上腺皮质激素、抗菌药杆菌肽、多粘菌素B等。20世纪70年代开始,合成的激素类药物开始出现,包括去氨加压素、性腺激素等。随着技术的发展,目前重组表达技术逐渐成为主流,包括甲状旁腺激素和胰高血糖素、杜拉鲁肽、胰岛素类似物等。已经有超过100种肽类药物上市。
多肽类药物是如何定义的呢?关于这点,其实是有一定争议的。通常,多肽药物氨基酸数量在3-100之间。一些非核糖体肽来源的分子,比如棘白菌素抗真菌药卡泊芬净、米卡芬净,脂糖肽类抗菌药万古霉素、达巴万星、特拉万星,以及其它天然产物衍生的非核糖体肽药物环保菌素、博来霉素等,FDA认为属于小分子类别。而用重组DNA技术表达的分子量更大的线性肽,如胰岛素类似物、甲状旁腺激素、人胰岛素生长因子、重组水蛭素等,虽然很多情况是由FDA CDER部门审评(生物药通常由CBER审评),但通常被视为生物药。2020年3月,FDA对肽类药物进行了分类,对于重组表达的超过40个氨基酸的分子被视为生物药物,按照BLA管理。对于小于40个氨基酸的分子(无论通过何种途径制备),或者通过化学合成制备的小于100个氨基酸多肽,被视为新分子实体,按照NDA途径申请。比如已经上市的GLP-1受体激动剂, Fc融合的度拉糖肽和白蛋白融合的阿必鲁肽氨基酸数量远超40个,按照BLA路径申报。艾塞那肽39个氨基酸组成,化合学成所得,按照NDA路径申报。
多肽药物在小分子和大分子之间的游走、不确定性,为这类药物的非临床评价带来很多挑战,是遵循化药的指南还是生物药的法规呢?所属类别不同,种属选择逻辑、免疫原性评估等方面均会有区别。
一般毒理
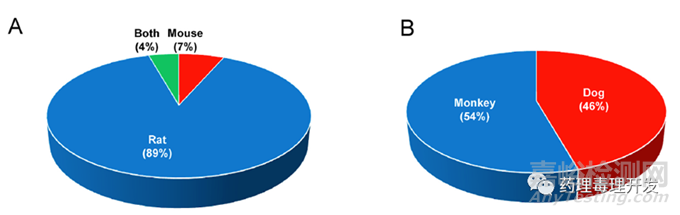
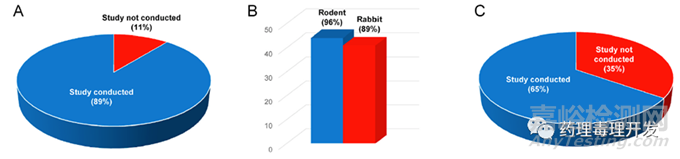
虽然ICH M3(R2)提及多肽药物不在该指南范围内,ICH S6(R1)则将化学合成或重组表达的多肽纳入指导范围,但实际情况是,几乎所有多肽类药物均参照了ICH M3(R2)开展啮齿类和非啮齿类毒理研究。不过,FDA综述资料里面并未提及如何选择的种属,从结果看,89%(41/46)啮齿类种属选择的大鼠,非啮齿类则比较均衡,46%(21/46)采用的犬,54%(25/46)选择的食蟹猴。多肽类药物的种属选择应该参照生物药思路更多一些。
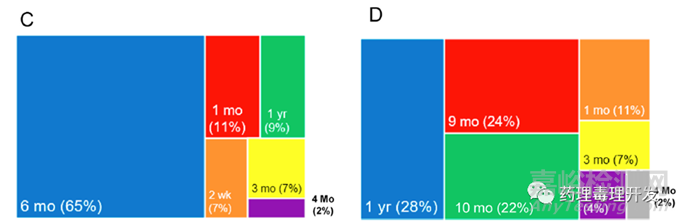
74%啮齿类研究最长给药周期持续超过6个月(C图),74%非啮齿类研究最长给药周期持续超过9个月(D图)。当然,这是最长给药周期,通常用于支持产品NDA/BLA,支持IND一般不需要开展如此长周期的研究,可根据临床试验方案和推进进度,分阶段开展。另外,大部分肽类药物适用于慢性病如糖尿病等治疗,用药周期长,超过3个月,所以最长给药周期基本是指导原则要求的上限。
生殖和发育毒性
几乎所有多肽药物均开展了Ⅰ段啮齿类生育力研究、Ⅱ段啮齿类或家兔胚胎-胎仔发育毒性研究和Ⅲ段啮齿类出生前、出生后发育毒性研究。具体比例参见下图。
含非蛋白质氨基酸的多肽
非蛋白质氨基酸(Non-Proteogenic Amino Acids, NPAA)是相对于组成蛋白质的20种常见氨基酸而言,指除组成蛋白质的20种常见氨基酸以外的含有氨基和羧基的化合物。有时为改善靶点亲和力或稳定性,会考虑引入非蛋白质氨基酸。NPAA既可以是天然氨基酸也可以是非天然氨基酸。21/47(45%)多肽类药物含有至少1个NPAAs,平均3.5个NPAAs。那么含有NPAAs的肽类药物需要额外开展毒理研究吗?大多数药物未被要求开展额外研究,哪怕使用的NPAA非常新,从未在多肽类药物中使用过。40多个多肽药物中,只有2例药物被FDA要求开展额外毒理研究,bivaluridine和icatibant。原因是这两个药物原液的杂质中均含有2个NPAAs,额外开展了重复给药毒性或细菌回复突变试验。
免疫原性测定
67%(21/46)的肽类药物进行了ADA检测,其中19/21开展了临床前和临床的ADA检测。比较有意思的是凡是临床前ADA阳性的药物,临床的ADA检测结果也是阳性的。反之则不成立,临床前阴性的,临床既有阳性也有阴性。ADA的产生与分子大小相关,产生ADA的肽类药物氨基酸数量为8-84个。不过,90%的肽类药物产生的ADA对药物的PK和药效没有影响。还有一个现象,76% ADA阳性的药物并未进行中和抗体检测。以上描述性统计结果可以看出,通常小分子不做ADA检测,生物药基本都会做,肽类药物介于二者之间,三分之二的药物开展了ADA检测。其实很多小肽免疫原性很低,即使添加佐剂,想获得抗药抗体都不太容易,所以肽类药物因免疫原性影响成药的风险较传统生物药低很多。但如果多肽分子量比较大,比如有几十个氨基酸,或者很多多肽药物做了长效化修饰,比如融合了人Fc、PEG等,或者不同靶点的多肽进行了融合,ADA的风险提高,评估起来就需要更加谨慎。
杂质研究
关于杂质问题,更多还是来自化药评价的思路。57%(16/28)的经化学合成、半合成的多肽药物被FDA要求开展单独的杂质毒理研究,包括体外遗传毒性或一般毒理研究。但关于多肽药物杂质问题,其实没有很明确的说法,能参考的指南还是ICH Q3A和ICH Q3B。
生物转化和PK研究
对于合成、半合成多肽,基本都开展了详细的生物转化研究,包括体外代谢、代谢物结构表征、代谢物体内表征等,类似化药的思路。
遗传毒性研究
通常生物药是不需要开展遗传毒性研究的。对于化药,细菌回复突变、染色体畸变及体内微核的遗传毒性组合试验是需要开展的。肽类药物虽然介于二者之间,但85%(39/46)的药物开展了遗传毒性相关试验。39个肽类药物的组成是:100%化学合成多肽(21/21)、61%(11/18)重组肽、100%(6/6)半合成肽、1个天然肽。不难看出,对于全合成、半合成肽、天然肽,通常需要开展遗传毒性研究,重组表达肽则要视情况而定,不是必须的。而且,全合成、半合成肽、天然肽开展的是完整的遗传毒性标准组合试验,11个开展遗传毒研究的重组肽中,仅7个开展了完整标准组合试验,剩余4个仅选择性开展了其中部分试验。39个多肽中仅2个出现阳性,而且是细菌回复突变阳性,染色体畸变阴性。这2个细菌回复突变阳性的多肽,glucagon和etelcalcetide,前者之后被证明是假阳性,后者经进一步体内外遗传学研究后,也判定为遗传毒性阴性。所以,39个开展遗传毒性研究的多肽,结果都是阴性的。
安全药理学
按照ICH S7B,49%(20/41)多肽药物开展了体外钾离子通道安全药理研究—hERG试验。大多数多肽药物开展了单独的大鼠和/或犬的心血管和呼吸系统安全药理学研究,个别采用非人灵长类进行的研究。
致癌试验
50%(23/46)的多肽药物开展了致癌性研究。其中,16/23开展了两个啮齿类种属的致癌试验,7/23仅开展了1个啮齿类种属的研究。不开展致癌试验的原因包括:1)临床给药周期短;2)肿瘤适应症;3)ADA的影响;4)有些作用机制产品已明确致癌风险,企业在说明书中直接注明致癌阳性,如GLP-1激动剂引发的甲状腺C细胞肿瘤。
13/23个开展致癌试验的肽类药物的结果是阳性的,其中7个作用于GLP-1靶点。前文已经提及,所有多肽药物的遗传毒性结果都是阴性的,故致癌阳性基本都是药物药理作用引起的。比如胰岛素具有促有丝分裂的作用,赖谷胰岛素和甘精胰岛素对啮齿类动物具有致癌作用。
总结
对于化药、生物药没有太大区别的试验,比如生殖毒性、安全药理、致癌,无论大分子还是小分子,需不需要开展,考量的因素是类似的。但对于ADA评价、遗传毒性、体外代谢、杂质研究、相关动物种属选择,就需要明确多肽药物的归属,到底是按照M3还是S6。由于缺少特定的针对多肽药物的指南,多数多肽类药物还是按照化药的思路开展非临床安评。没有统一的指南,就很难有统一的行业操作标准,大家也是根据自己的理解、过往产品的研究经验,化药和生物药交叉结合的思路设计非临床试验。已经上市的多肽类药物很多,可以根据自己产品的特点比如靶点、作用机制、适应症等,结合已上市药物的经验,必要时跟监管机构沟通,合理设计非临床研究策略。