您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-08-13 17:21
NO.1 治疗器械能量关系研究指南?
答:这是非常专业的问题,需要根据具体的产品确定,欧盟没有专门针对具体产品的指南,可以参照美国FDA的指南,如:General Considerations for Animal Studies Intended to Evaluate Medical Devices,Reporting of Computational Modeling Studies in Medical Device Submissions等。
NO.2 SOTA检索要求和关键词选取?
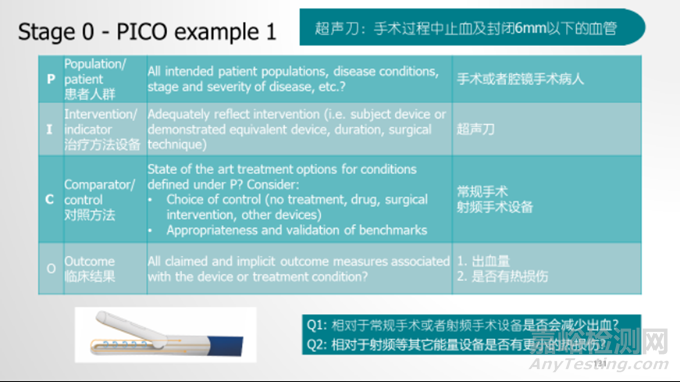
答:根据MDR 附录XIV,临床评价计划要基于SOTA建指示清单和规格参数(an indicative list and specification of parameters based on the state of the art)。SOTA指标的建立可通过检索来建立,参照MEDDEV 2.7.1 Rev4,常规包括互联网检索和文献检索。互联网检索包括临床指南的检索,帮助确定各种治疗方案和替代技术等,明确公认的SOTA技术。文件检索可确定各种治疗方案的具体的指标。大家使用比较多的检索策略是PICO,P是指患者人群和适应症,可以作为关键词选择,I是指自己器械的治疗方法,C是指对照,因为SOTA的确认要考虑所有治疗方法,故此I和C相关治疗方法,以及具体器械的名称都可以作为关键词进行选择。C是指临床结果,可以不作为关键词选择,以免限制捜索的范围和结果。
NO.3 三类治疗医疗器械临床试验样本需求和国内临床试验数据可否用于欧盟临床评价?
答:临床试验的样本量的确定是一个非常专业的问题,多根据统计学的方法由统计专家确定。中国的临床试验应该要包括在CE认证的临床数据内,因为欧盟MDR法规要求的临床评价要包括所有的好和不好的数据,并且临床试验的数据是最高水平等级的数据,理应包括。
NO.4 临床评价中,产品材料没有完全等同的情况下,需要补充什么资料(或补充什么测试),以符合要求。
答:按照MDR关于材料的等同,使用了相同(Same),而不是类似(Similar),并且MDCG 2020-5将其和MEDDEV 2.7.1第四版进行了对比,故此,如果进入人体的材料是不同的,这种情况在MDR下是不能等同的。如果是要提供证实材料等同的证据,可以参照MDCG 2023-7指南附录II(Appendix II: Hierarchy of levels of access to the data),包括签署协议,或者如您提到的对比测试等。
NO.5 临床评价哪种临床评价路径可以免临床试验。
答:MDR Article 61和Annex XIV讲述了临床评价的要求,按照MDR Annex XIV第3条款,以及MDCG 2020-5指南等同可以建立,或者可以按照Article 61第10条走不需要临床数据的路径,就可能不需要临床试验。但是,针对三类和植入的器械,MDR Article 61第4条要求必须要进行临床试验,除非满足Article 61第5条(不同制造商之间等同需要有合同)和第6条的要求(Legacy产品和WET产品可能豁免临床试验)。
NO.6 上市后监督如何更好地设计和执行?
答:上市监督计划按照MDR附录III的要求进行,考虑1.1(a)所有收集到的信息,包括严重不良事件、不严重的时间、非预期的副作用、客户反馈和投诉、以及类似器械的信息等。以及1.1(b)条款所要求的主动的系统的流程……
NO.7 临床评价和上市后临床跟踪的关系是什么?
答:上市后临床跟组PMCF主要是跟踪器械的长效安全性能、剩余风险和不确定的事项等,它会根据临床评价的输出结果而定,具体的可以参照MDCG 2020-7,以及MEDDEV 2.12/2 Rev.2 指南。
NO.8 所有变更都要提前与公告机构沟通吗?
答:MDR法规Annex IX第2.4条款要求制造商报告体系的重大的变更给公告机构(Annex IX 2.4. The manufacturer in question shall inform the notified body which approved the quality management system of any plan for substantial changes to the quality management system, or the device-range covered. The notified body shall assess the changes proposed, determine the need for additional audits)。
MDR法规附录IX第4.10条要求,针对产品证书,制造商需要报告并获得公告机构批准可能影响器械安全和性能以及使用条件的变更(Annex IX section 4.2: Changes to the approved device shall require approval from the notified body which issued the EU technical documentation assessment certificate where such changes could affect the safety and performance of the device or the conditions prescribed for use the device.)。
MDR法规Article 120 Transitional provisions 还要求宽限期(Grace Period)内不能有重大的设计和预期用途的变更(The ‘grace period’ requires that there are no significant changes in design and intended use.)。
具体的可以参照NBOG 2014-03,以及MDCG 2020-3 Rev.1指南。
NO.9 经济运营商是否要纳入采购的合格供应商名录?建立供应商档案?
答:MDR法规增加了经济运营商的法律职责,制造商将经济运营商选择和控制建立流程,或作为供应商来进行控制,是一个比较好的实践。
NO.10 有宽泛适用症的成像设备(比如通用的x射线影像设备),和专用于某个适应症的成像设备(比如乳腺x射线机),在定义临床受益的临床结果时是否要有不一样的考虑?比如前者可以考虑成像质量,后者要考虑准确性?还是说两者都可以考虑成像质量?
答:临床受益根据预期用途和宣称(claim)而定,具体的可以参考文献中的受益终点和指标,对于广泛的临床受益,可以考虑使用他们达到预期的功能和性能来表示他们的临床受益,或者使用有效的替代终点(surrogate endpoints),并说明理由。可参考MEDDEV 2.7.1 Rev.4。
NO.11 保险问题,制造商没有买保险,公告机构审核通过发证了,就可以在欧盟销售了吗?或者说,没有保险,只要有证就可以卖。
答:按照MDR Article 10制造商的法律职责,制造商需求按照Directive 85/374/EEC,对其产品所承担的可能的法律责任(potential liability),提供充分的财务覆盖(sufficient financial coverage),保险是多数制造商选择的一种方式。具体的审核,有些公告机构会确认是否有保险,有些公告机构会认为是制造商自己的法律责任,审核过程中不关注是否有保险。具体的和您的公告机构确认。
NO.12 公告机构对检测报告的要求是什么?自检报告可以吗?有没有像国内注册有cma证书资质的要求?
答:不同的公告机构对采用标准的检测报告有不同的要求,有的公告机构需要自己检测,有的可以认可有相关资质实验室的检测报告,如ISO 17025认可,具体的和您的公告机构咨询。
对于设计输入和产品规格的很多检测项目,很多是企业自己来进行检测的,包括需要考虑合适的检测方案和方法,基于统计学的样本理由,以及测试人员、测试设备等的追溯。
NO.13 CECP审核需要制造商自己发起还是公告机构去完成?
答:按照MDR Article 54、Article 55,以及Annex IX 第5.1条款,CECP是针对III类植入器械,以及IIb有源输注类器械,在公告机构审核完成之后,将其临床评价审核报告(CEAR),以及制造商临床相关的文件,转交欧盟委员会的专家团队(Expert panel)进行CECP流程。
NO.14 法规61.10条款怎么解读?是指可以走性能评价路径么?
答:MDR Article 61(10)是指不需要临床数据去支持满足GSPR要求的器械,通过性能测试、台架测试的证据去支持。通常是指低风险、不和人体接触,没有临床受益宣称的器械。但也需要完成临床评价报告。
NO.15 PMCF可以作为临床评价缺失部分的补充么?
答:PMCF不能豁免或者降低上市前所需要的临床证据,但对于长效安全、罕见证等可能可以通过PMCF的数据去跟进和补充。可参照法规、MDCG 2020-7,以及MEDDEV 2.12/2 Rev.2 指南。
NO.16 如我司和公告机构对于分类有歧义,需要向中国药监局申请仲裁?
答:按照MDR Article 51 第2条款,如果制造商和公告机构对分类有分歧,可以向制造商或者其欧代所在国的主管当局申请分类的仲裁。
NO.17 目前WET产品是否只认article里面的清单?
答:Article 61第6条(b)针对成熟技术(WET)的豁免临床试验的产品清单,是法规的要求,如果需要修订,应该需要通过法律修订而进行。另外,MDCG 2020-6有WET的定义和解读,有时WET也理解成Standard of Care产品,这个和WET清单是不同的,大家可以参照。
NO.18 受益风险比的分析,是否要每个受益配对每个风险来分析?
答:受益和风险通常是在不同维度的考虑,有时很难配对,多数情况下是针对SOTA进行衡量。

来源:Internet


