您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-08-22 08:11
|
NDA 序号 |
216059,批准时间:2023年 |
|
申请人 |
Loxo 公司,礼来子公司 |
|
药物名 |
JAYPIRCATM (pirtobrutinib) 片 |
|
制型 |
片剂 |
|
规格 |
50mg和100mg |
|
途径 |
口服 |
|
最大日用剂量 |
200mg |
|
原研/非处方 |
原研 |
|
适应症 |
既往接受过共价BTK抑制剂治疗的成年套细胞淋巴瘤(MCL)患者的治疗 |
|
外观 |
Pirtobrutinib片50 mg或100 mg薄膜包衣、凹口片剂,用于口服给药。50mg片是一种蓝色的弧形三角形片剂,一边是“50”上的“Lilly ”,另一边是“6902”。100mg片是蓝色的圆形药片,一边是“Lilly ”,另一边是“7026”。 |
|
储藏条件 |
储存于20°C-25°C(68°F-77°F);允许在15°C和30°C(59°F和86°F)之间偏移 |
生物药剂学评估:拟议的QC溶解方法(USP方法/桨法,75rpm,900毫升pH 6.8(XX)缓冲液;37 C)和修订后的溶出度标准(Q =(30分钟XX%)适用于pirtobrutinib片剂在批量释放和稳定性测试/保质期期间的溶出测试。请注意,提交的PBBM的基于生理的药代动力学模型(PBPK)被认为不足以支持申请人最初提出的(更宽泛的)溶出度接受标准。
有足够的体外和体内数据(i)支持将最终拟议上市的药物产品(50mg和100mg)与关键临床试验(100毫克)和注册/稳定性研究(50mg和100mg)中评估的片剂批次的桥接,以及根据21 CFR 320.22(d)批准申请人对拟议药物产品50毫克强度的生物豁免请求。
在豁免50mg,申请人递交了如下14个文件,以支持将50mg规格的上市申请。

基于pirtobrutinib药物在37C下各种pH介质和生物相关介质中的pH-溶解度数据,根据BCS分类标准,pirtobrutinib可以归为低溶解度化合物。后面更多内容被隐藏。
原料药渗透性:高渗透
申请人补充的报告研究LOXO-BTK-20007/第2部分,pirtobrutinib(两片100毫克“T2”pirtobrutinib片剂服用;关键临床试验批次19-0216-2)的绝对生物利用度(Fpo)为85.5%(相对于静脉注射(6)含有放射性标记药物物质的溶液,Fpo>85%表明pirtobrutinib可被认为是一种高渗透性药物物质。另外,企业没有提供使用Caco-2模型的体外渗透数据。根据相对溶解度和动物相对生物利用度数据,预计pirtobrutinib片在人中的口服生物利用度会更高。
制剂溶出:在生理pH值范围内由缓慢到非常迅速。
Pirtobrutinib片为速释药物,其溶解度与pH值有关,在各种pH介质中显示溶解缓慢,除了在pH 6.8(b)*缓冲液中,有关详细信息,请参阅资料中表3.2中各种pH值和生物相关介质中“T2”片剂(100毫克开发批次BREC-2224-035)的溶解度数据。P.2.2.1.5.3.1.3-1,以及图3.2。P.2.2.3.1.1-1。另请参阅图3.2。P.2.2.3.2.2-1至图3.2。P.2.2.3.2.2-3,用于100mg注册稳定性批次D345037的pH溶解数据。50mg规格的T2片剂也观察到类似的pH溶解趋势。
「解读」pirtobrutinib属pH依赖性化合物。FDA同意了企业拟定的溶出方法,但没有同意企业基于PBPK模型申请的宽泛溶出标准。溶出标准不仅涉及PK,还对于批次的放行起到区分作用,能将不满足质量要求的批次剔除出去。
企业申请了两个上市规格50mg和100mg,其中关键临床使用过100mg规格,50mg没有用于关键临床,仅用在其他临床研究,这种情况下50mg可以申请上市吗?
基于FDA给到的审评结果,企业申请了生物豁免,提交了50mg的其他临床数据,以及50mg规格与100mg规格之间的对比数据等,最后FDA同意了企业的豁免申请。
B.溶出方法和接收标准
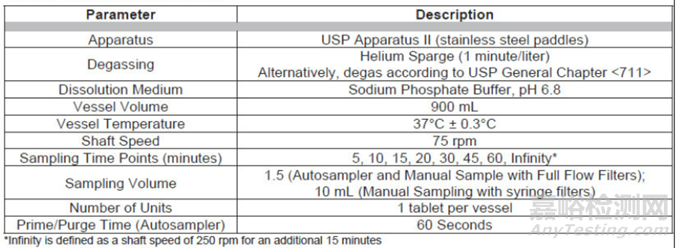
pH值为6.8缓冲盐被选为最终的QC溶出介质,因为在pH值为6.8条下化合物可实现3倍漏槽条件和完全溶解,此外所选的介质足以实现溶出方法区分能力,改变药物的质量属性,该溶出方法足以区分。
为降低溶出数据的波动,选择了浆法2,75转速度,900mL的溶出介质。
漏槽条件:相关描述被隐藏了。
区分力:只提到了原料药的粒度部分,其它内容被隐藏了。从以上数据来看,拟定的溶出方法具有一定的区分力。此外,拟定的溶出方法能够产生预期的水平等级关系,相关具体描述被隐藏了。
溶出方法验证:溶出方法验证了HPLC与紫外检测。研究了分析方法的线性、准确性、精度、范围、耐用性(关于溶解法和HPLC方法参数)、标准和样品解决方案的稳定性、过滤器兼容性(全流速过滤器,采样类型(自动或手动))。
「解读」本品是常规速释固体片剂,其溶出是制剂的一个关键质量属性,因此在相关的P2.3制剂开发部分,需要详细的溶出方法开发内容。审评报告只重点摘出了很少一部分论述,其中溶出的区分力被重点提及。提及了对原料药粒度进行了考察,溶出方法可一定程度区分不同粒度原料药得到的片剂。具体研究数据未在审评报告中展示。
溶出接受标准,进行了修订。
(最初)拟定的溶出接受标准是“Q =30分钟XX%。
根据拟定的QC溶出度方法(方法G2535)生成的四个关键临床试验批次(即100mg T2片剂使用在支持稳定性研究中批次D322845和D322846,以及100mgT2片剂批次使用在初级稳定性研究D345038和D345039)的溶出度表征数据,FDA建议企业收紧溶出度接受标准,以“Q =30分钟XX%。
收紧的溶出标准(Q =XX)在考虑其他三个额外关键临床试验100mg“T2”片剂的快速到非常快速的溶解特性时(由倒数第二个QC溶解方法生成),FDA认为,使用最终的QC溶解方法可以实现类似的(如果不是更快的)溶出。
根据使用倒数第二种和最终QC溶出方法为一个研发批次(即100mgBREC-2224-035)生成的溶出曲线数据的比较,两种方法的溶出曲线相似,最终方法G2535预计将产生比倒数第二种方法BTP-548略高的溶出数据(即在30分钟时,(*%更高的溶出值))。因此,对于任何给定的药物批次,同一溶出采样时间点(例如30分钟)的溶出数据预计最终溶出方法产生的溶出数据略高于倒数第二个溶出方法产生的溶出数据。因此,基于使用倒数第二和最终的溶出方法,FDA认为,任何使用倒数第二个溶出方法在30分钟内都能达到用最终QC溶出方法在30分钟内达到的Q值。
此外,对于Q=XX%30分钟时,将足以拒绝那些不符合质量接受标准的片剂批次,这些批次包括那些明显低于目标溶出的片剂。FDA认为,进一步评估单点溶出标准区分批次的能力很重要。
此外,即使根据BCS分类标准,pirtobrutinib化合物是一种低溶解化合物,基于以下考虑,制定2点溶出标准被认为是没有必要(即防止超快速溶解,这可能导致更短的Tmax和更高的Cmax),基于以下考虑:
1. 基于企业递交的数据,一些关键临床试验批次表现出非常快速的溶出(即在15分钟内至少85%)。
2.注册批次和工艺验证批次在12个月的长期稳定性时间点中,在QC介质中都表现出快速的溶出释放(15人种内至少85%)。
3.一项彻底的QT研究显示,超治疗剂量(即900mg)的pirtobrutinib片没有导致健康受试者的临床相关QT延长。
4.根据对MCL患者疗效和安全性的暴露反应分析,pirtobrutinib片不是一种治疗窄窗药物。
「解读」针对pirtobrutinib制剂的溶出接受标准,FDA从以下几个方面给到意见:
1. 建议收紧溶出接受标准,至30分钟XX%,理由是基于多个关键临床批次的溶出数据,展示了药物在QC介质条件下非常快速的溶出特征。
2. 基于递交的溶出数据,使用修订后的按受标准可以区分拒绝那些不满足质量标准的批次。
3. 即使属于BCS 2/4的低溶解化合物,也可以不设定2点溶出接受标准,FDA给到了非常具体的4点考量。即关键临床批次的快速溶出表现、注册批次/工艺验证批次在稳定性研究期间的快速溶出、超剂量时没有QT的延长,以及药物本身不属于治疗窄窗药。
4. 溶出方法的选定非常重要,最终会决定接受标准时间点和Q值的选择,以及是否设定2点的接受标准。
5.企业在溶出方法开发过程中,一些关键临床批次的测试有使用到另外一个溶出方法测试而不是最终的QC方法测试,两种方法差异不大,最终的QC方法是在后期拟定的,溶出可能更快,前期的部分批次可能没有被QC方法测试,这种情况下,FDA接受了企业的做法。
稳定性批次的溶出数据
50mg和100mg规在HDPE瓶中的三批注册稳定性批次的12个月长期(30°C/65% RH)和6个月的加速(40°C/75% RH)稳定性数据,在拟定的包装中储存,片剂最初拟定的有效期为24个月。值得注意的是,最终拟定的QC溶出方法(方法G2535)用于分析注册批次和其它稳定性批次的稳定性样品所有溶出曲线数据。在12个月的长期稳定性研究期间,30分钟的溶出数据没有观察到有稳定性趋势。在加速稳定性条件下第6个月,两种规格片剂的溶出度似乎都略有下降;然而数据仍在拟定修改的限度耐受范围内(Q = XX%在30分钟),因此FDA建议企业收紧溶出接受标准在该Q值。
注意到,100mg规格三个注册稳定性批次的两批次(即D345038 A/C和D345039 A/C)在稳定性进行到10个月至11个月时,用于关键临床试验。图5显示,这两批稳定性批次/关键临床批次没有出现各稳定性时间点的溶出数据变化。另外两个关键临床试验批次(D322845、D322846)在关键临床试验使用时,其稳定性时间点为7个月至11个月。
在企业回复FDA中,企业(i)同意FDA的建议,基于关键临床试验批次、注册批次/稳定性批次和工艺验证批次的溶出数据,以及FDA对PBBM模型数据的评估,将溶出接受标准从“Q= XX%,30min”收紧到“Q = XX%,30 min”,(ii)修订成品QC溶出接受标准和其他相关的NDA文件,以及(iii)确认,根据迄今为止得到的稳定性溶出数据(以及药品稳定性证据),注册批次能够相应地符合(并预计能够)符合修订后的溶出接受收标准。
「解读」FDA从另外一个维度,即关键临床批次和注册稳定性批次在稳定性时间点及用药时间点,其溶出数据仍然符合修订的接受标准。企业QC溶出接受标准不仅基于多批次的关键临床批次,注册批次的溶出曲线数据,同时基于3批注册批次在稳定性研究阶段各时间点的溶出数据情况。本制剂同时也收集了PPQ批次的溶出数据,进一步证实了商业化批次溶出数据仍可满足修订后的溶出标准。

来源:Internet


