您当前的位置:检测资讯 > 实验管理
嘉峪检测网 2024-10-21 18:17
什么是琼脂糖凝胶电泳?
琼脂糖凝胶电泳是一种电泳形式,用于根据核酸(DNA或RNA)片段的大小进行分离。当施加电流时,带负电的DNA/RNA通过琼脂糖凝胶的孔隙向凝胶带正电的一端迁移,较小的片段迁移较快。由此产生的条带可以用紫外线(UV)光来观察。
琼脂糖凝胶电泳的工作原理是怎样的?
琼脂糖是琼脂的一种成分,它形成了一个由螺旋状的琼脂糖分子组成的三维凝胶基质,这些螺旋状的琼脂糖分子在超线圈束中被氢键固定,有通道和孔隙,分子能够通过。当加热时,这些氢键断裂,使琼脂糖变成液体,并允许它在复位前被倒入模具。
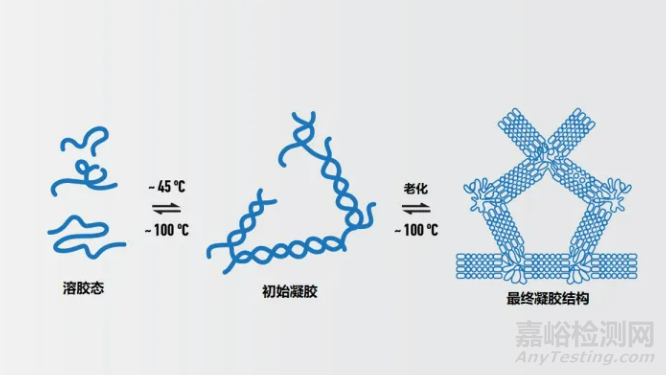
琼脂糖在凝胶中的百分比影响了孔的大小,从而影响了可能通过的分子的大小以及通过的速度。琼脂糖的百分比越高,孔径越小,因此能够通过的分子越小,迁移速度越慢。
在分子生物学实验室中,0.7~1%的琼脂糖凝胶通常用于日常的DNA分离,对0.2~10kb范围内的片段提供良好、清晰的区分。较大的片段可以用较低百分比的凝胶来解决,但它们会变得非常脆弱且难以处理,而较高百分比的凝胶会对小片段提供更好的分辨率,但很脆且可能凝固不均匀。
由于DNA在肉眼中是不可见的,在凝固过程中,一种夹层染料如溴化乙锭(EtBr)被纳入凝胶中。这与DNA结合,并在紫外线下发出荧光,从而使DNA片段可以被观察到。存在的DNA越多,条带就越亮。
与加载染料混合的样本被放置在凝胶的一端,凝胶被浸泡在运行缓冲液中。然后电流通过凝胶槽两端的电极穿过凝胶。
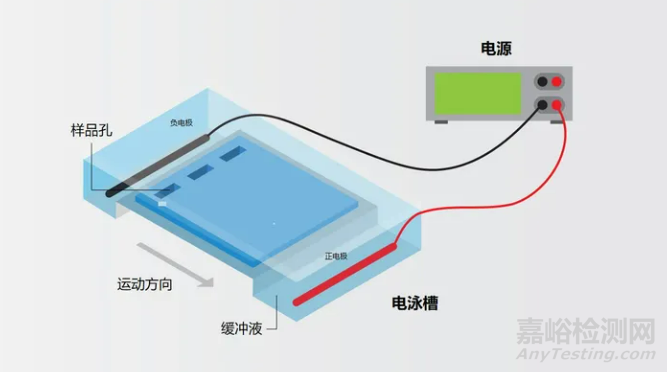
琼脂糖凝胶放置在缓冲液槽中,样品与加载染料混合后放置在凝胶一端的孔中,施加电流使带负电的DNA向正电极(阴极)移动
当样品运行得足够远以获得足够的分离时,将凝胶从槽中取出并放在一个紫外光箱上。然后,夹层染料可以使样品条带可视化,并通过与已知条带大小的DNA marker进行比较来确定其大小。迁移距离和片段大小之间的关系是非线性的,增加了包括尺寸标记作为指导的重要性(图3)。
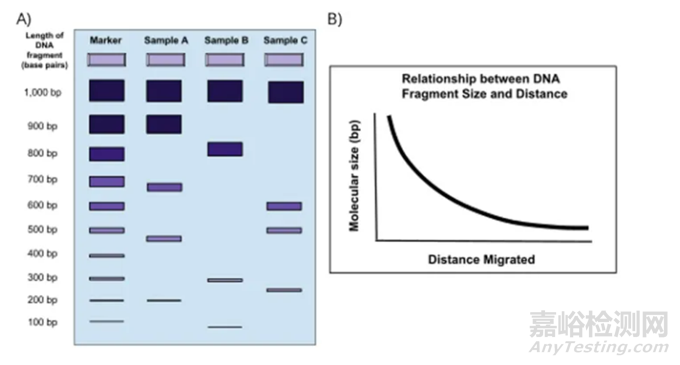
A)图片左侧描述了DNA电泳的典型结果。在左边,有一个尺寸标记,用来作为样品DNA片段长度(以碱基对为单位)的参考。标记的右边是三个样品。图片显示了较小的DNA片段如何在琼脂糖凝胶中比较大的DNA片段移动得更远。
B) 图片右侧的图表显示了DNA片段的大小与迁移距离之间的非线性关系。这是一条负的曲线,当DNA片段变大时,它们在凝胶中迁移的距离就会减少。
DNA凝胶电泳的步骤
在选择、设置、运行和分析琼脂糖凝胶的过程中,有一些关键步骤,我们现在将介绍这些步骤。
确定所需的凝胶百分比
0.7-1%的琼脂糖凝胶通常对大多数应用来说是足够的,但重要的是选择一个适合你的样品和预期片段大小的百分比。将琼脂糖粉末与用于运行凝胶的相同类型的缓冲液结合起来,加热使混合物融化,避免沸腾。
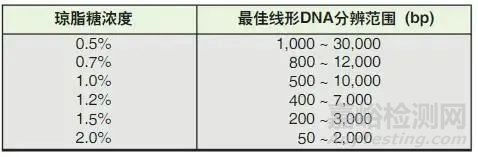
下表为不同琼脂糖凝胶浓度所对应的线性DNA最佳分辨范围:
三乙酰-乙二胺四乙酸(EDTA)(TAE)或三硼酸-EDTA(TBE)[5]通常是首选的缓冲液,因为三酸溶液是微碱性条件下的有效缓冲液,可保持DNA去质子化并溶于水。EDTA是一种螯合剂,能使可能损害被分析的DNA的核酸酶失活。
凝胶浇注
选择所需尺寸的凝胶浇注模具和梳子,为所有样品和marker提供足够数量的孔,以及容纳每个待装样品数量的孔容量。用铸造框架或胶带固定模具的两端,以便在凝胶凝固时将其固定。
在模具的底部加入DNA夹层染料,如果使用的是EtBr,通常浓度为0.2-0.5 µg/ml。EtBr是一种诱变剂的证据目前仍有争议,但因此,许多实验室已经转而使用替代品[6],如GelRed。
加入凝胶,注意不要过度填充模具,并确保夹层染料均匀混合。凝胶太热时不要倒入,否则模具可能会变形或破裂。
将样品/marker与加载染料混合
加载染料具有多种功能,它们可以让用户看到他们原本无色的样品在哪里,使其更容易将样品准确地移入孔中,从而减少孔间样品交叉污染的可能性。
当凝胶运行时,染料与样品一起迁移,使用户能够知道样品在凝胶中的位置,并防止样品跑得太远而流失到缓冲液中。没有加载染料的DNA样品在加载时也会倾向于分散到运行缓冲液中,因为它们的密度较小。
因此,大多数加载染料含有甘油或Ficoll,这使得样品-染料混合物更密,所以它沉淀在孔的底部。溴酚蓝是一种流行的着色剂选择,但有些也含有额外的染料,如二甲苯氰醇。虽然可以购买加载染料,但许多实验室选择自己制作。
如果你的样品量非常小(例如,少于5 µL),在这个阶段加入一点水可能是有利的,这样更容易有效和均匀地装载到凝胶孔。
同样,如果你预计某些样品中的DNA浓度比其他样品高得多,那么在这个阶段也可能有必要向这些浓缩样品的样品-染料混合物中加水。
如果你不这样做,在可视化过程中,这些条带给出的强信号可能会掩盖较弱的条带,或者需要对强条带进行过度曝光以观察较弱的条带,在凝胶图像上形成明亮、扭曲的区域。
凝胶装入
从设定好的凝胶上取下浇注框/胶带,并将其放入凝胶槽中,确保孔位于负电极(一般为黑色),将运行缓冲液(TAE或TBE)注入槽中,使凝胶被淹没。
小心地取下梳子,轻轻地将样品-染料(如果使用水)混合液移入孔中。尽量避免用移液器吸头接触孔的边缘,因为它们可能会破裂,使一个样品跑到下一个孔中。过量的孔会产生同样的结果。DNA的样本量过大也会在运行过程中减慢DNA的迁移。
装上标记marker,最好是在样品行的两端各装一个。凝胶不一定总是在一条完美的直线上运行,所以在两端各装一个marker可以更容易确定存在的片段的大小。有多种marker可供选择,并且标明了不同的尺寸,选择一个最适合你期望的尺寸。
凝胶运行
将盖子放在电极黑对黑、红对红的罐子上,并将电极插入一个电源盒,也是黑对黑、红对红,这与凝胶罐一起构成了凝胶电泳仪。
确保电极和盖子的方向正确,否则你的样品会从孔中倒流到运行缓冲液中。设定凝胶运行的时间和电压,120V、35分钟是一个很好的近似值,但是这应该根据所使用的凝胶比例和预期分离的片段大小进行调整,以获得良好的电分离。
在琼脂糖凝胶上施加电流会使其发热,电压越高,发热越多,所以当运行低百分比的凝胶时,最好使用较低的电压以防止熔化。
增加电压以使凝胶运行得更快是很诱人的,然而,这可能会导致【笑脸凝胶】,即带子在两端向上弯曲,使其难以确定正确的带子大小。这是指凝胶开始轻微融化,使条带运行不均匀。这也会导致条带出现涂抹状和不清晰的情况。
可视化
一旦样品在凝胶上运行了大部分(染料前部会使其可见),关闭电源。
戴上手套,轻轻地将模具中的凝胶从槽中取出,排掉多余的运行缓冲液,并将其转移到一个适当的容器中的紫外箱中,以便进行可视化。更换手套以防止凝胶或运行缓冲液中的夹层染料污染周围的表面、门把手等。
如果下游应用需要DNA片段,可以用手术刀小心地从凝胶中切除相应的条带,同时将其放在黑暗房间的紫外光箱上。确保你戴上紫外线面罩,并在灯箱开启时保持皮肤覆盖,以防止紫外线对皮肤或眼睛的伤害。
凝胶电泳结果的判读
琼脂糖凝胶可以在暗室中的紫外光箱上进行观察,或者使用与相机相连的独立灯箱。
无论使用哪种系统,紫外光从下面照射凝胶,DNA条带由于与它们结合的夹层染料而发出荧光,可以用带有专门的紫外线过滤器的照相机来记录。
标记物marker有一份说明书,表明它们包括的每个条带的大小。通过将其与样品通道中的条带进行比较,可以确定条带的大小。样品之间DNA的相对数量也可以进行比较,因为较高的DNA浓度会产生较亮的条带。图中显示了一个例子。
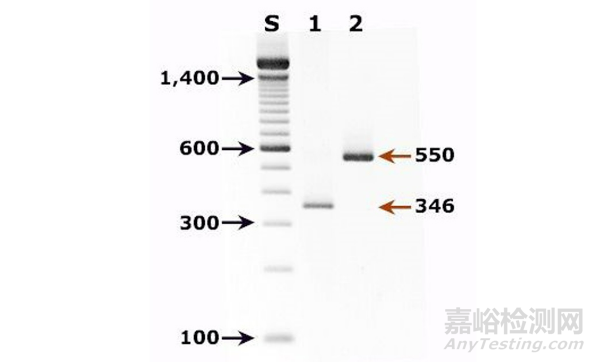
琼脂糖凝胶电泳的技巧
胶液的制备
称取0.4g琼脂糖,置于200ml锥形瓶中,加入50ml0.5×TBE 稀释缓冲液,放入微波炉里加热,直到琼脂糖全部熔化,取出摇匀,此为0.8%琼脂糖凝胶液。总液体量不宜超过锥瓶的50%容量。否则会溢出来的。加热过程中要不时摇动,使附于瓶壁上的琼脂糖颗粒进入溶液。加热时应盖上封口膜,以减少水分蒸发。
胶板的制备
倒胶时的温度不可太低,否则凝固不均匀;
速度也不可太快,否则容易出现气泡;
待胶完全凝固后拨出梳子,注意不要损伤梳底部的凝胶。
加样
注意上样时小心操作,避免损坏凝胶或将样品槽底部凝胶刺穿。
电泳
加完样后,合上电泳槽盖,立即接通电源。控制电压保持在60-80V,电流在40mA以上。当溴酚蓝条带移动到距凝胶前沿约2cm时,停止电泳。
染色
未加EB的胶板在电泳完毕后移入0.5μg/ml 的EB 溶液中,室温下染色20-25 分钟。
观察和拍照
在波长为254nm 的长波长紫外灯下观察染色后的或已加有EB 的电泳胶板。DNA 存在处显示出肉眼可辨的桔红色荧光条带。紫光灯下观察时应戴上防护眼镜或有机玻璃面罩,以免损伤眼睛。
琼脂糖凝胶电泳问题点及分析
加样孔有荧光
下图的红框部分就是一个典型的加样孔有荧光的例子。发生这种现象可能的原因如下:

首先一定有DNA 的滞留,其次多含蛋白质残留;如果是比较特殊的样品,其杂质也可能导致荧光。蛋白质几乎不会被EB 染色,能出现荧光,则一定含有核酸。非常巨大的基因组DNA (>50kb),如果使用普通的琼脂糖电泳,往往不能电泳出孔,单独就可能滞留在加样孔中产生荧光。
更多的时候,是比较大的DNA 与残留的蛋白质结合后,电泳不能出孔而在孔中产生荧光。酶反应产物,如果没有经过纯化去除酶,也非常容易在孔中出现荧光,其原因是酶与核酸几乎都有比较强的结合能力(基因组DNA 的PCR 产物电泳往往在孔中都有荧光,而且荧光强度与体系的特异性成反比。)。如果是植物样品,还可能由其它杂质残留引起。
那么,这么多种可能性,到底是什么东西残留呢?
按照经验是:蛋白质残留的荧光有点发闷,其它杂质残留亮得很刺眼,且薄得锐利。如上面那个例子,这么亮,这么锐利,肯定不会是蛋白质啦。
总RNA 非变性电泳显示28S 不如18S 亮
如下图所示,18S 明显地比28S 亮了。
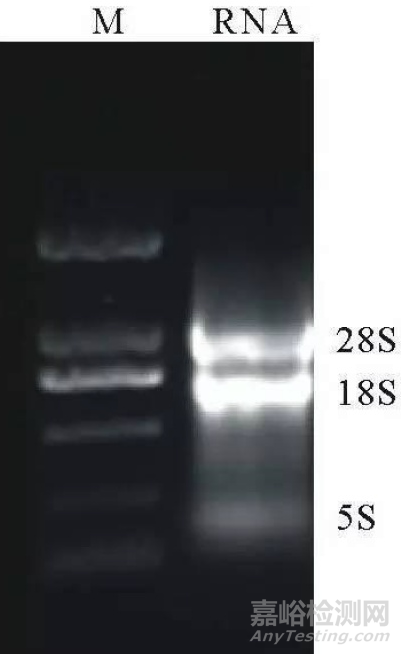
如果28S 和18S 的条带清晰无弥散,那么多提示28S 没有被EB 饱和。RNA 残留多时,基因组DNA 的电泳也会有相似现象。
解决方法:简单的补染就可以解决此问题。再次电泳时,或者降低上样量,或者直接增加胶中EB 的量。保证28S 比18S 亮堂很多很多。
降解
降解的现象,情况是比较多的总结如下:主带不再突出,向小片段方向发生弥散,亮度比较均衡地衰减。
如果同时还伴随下列的一个或者多个现象,则需要更进一步的检测:加样孔非常的亮、弥散发生在主条带位置的上下两个方向、从加样孔即开始发生弥散。
如下图所示,红框里面的就是典型的降解的现象。
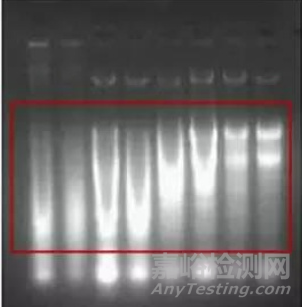
其实,以现在的裂解液的裂解能力而言,降解的发生主要是在彻底匀浆之前,只有极少量由不干净的溶解液导致。
以总RNA 抽提为例,总RNA 的质量由高到低依次为悬浮细胞、贴壁细胞、组织。彻底裂解悬浮细胞的时间最短,彻底裂解组织的时间最长,这就是降解多发生在彻底匀浆之前的一个佐证。再看一看新鲜样品和冷冻保存样品,非常严格的冷冻保存和匀浆操作的确可以确保冷冻样品的核酸的质量;但是,有许多实验室并不具备严格的保存手段,实验人员的操作也并非完美,其结果就是核酸的降解。
事实上,RNA 抽提受的影响因素太多,就以基因组DNA 的抽提为例看一看吧。
假定消化使用的是含蛋白酶K的溶液,无论你使用的是新鲜样品还是冷冻保存样品,如果混匀彻底,可以预期的降解为:细胞:不应该降解,碾碎的组织:不应该降解,大块组织:部分降解。
如果电泳发现新鲜的细胞和碾碎的新鲜组织发生了降解现象,该现象是假象;如果电泳发现冷冻的细胞和碾碎的冷冻组织发生了降解现象,该现象不是假象,就是样品在保存中已经降解了。说得更极端一点,即使蛋白酶K失活了,消化试剂的裂解能力也足以保证细胞的基因组DNA 在抽提过程中间不被降解。
那么,如何才能减少或者杜绝核酸降解呢?
那就是:一定要将重点放在样品被彻底匀浆之前,其次就是要缩短样品从脱离原来的生存环境或者低温到被彻底匀浆之间的时间,越快越好。

来源:Internet


