您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-11-26 17:43
今天跟大家介绍的是在整个项目中可行性阶段,作为这个项目的注册工程师要做哪些工作呢?
-可行性阶段,我们要做什么?-
从公司有一个新项目新产品的想法开始,那一定是先进行这个项目的可行性调研,在这个时候,作为这个项目的注册工程师,你就要上岗啦!!!
这个时候,我们能做什么呢?
1,确定产品的分类和分类编码
对于一些有明确分类的产品,在医疗器械分类目录中就可以找到啦,那对于一些不太明确分类的产品,我们就要申请分类界定(对于分类界定,请关注公众号后面会跟大家分享哒 ),最终以药监界定的分类结果为准。
2,明确产品的产品名称、结构组成和适用范围。
很多小伙伴要说了,确认产品名称、结构组成和适用范围那不是技术的职责吗?
实际上技术部给出的一些信息是不符合法规或指南要求的,所以在这个过程中,RA也要协助技术部门、市场团队共同协商一起决定的!
有些产品的结构组成和适用范围会在分类目录中明确,也有些产品在产品注册审查指导原则中也有规定,除此以外对于没有很明确的产品就要调研一下竞品的描述、以及销售端在入院时医院有什么特殊描述的要求?综合考虑确定产品名称、结构组成和适用范围。
这样做的目的是为了:
避免产品在注册申报阶段,因为产品名称、适用范围、结构组成描述不规范的问题被审评要求重新修改,到那时对于这个项目的整套文件及报告进行修改,任务量是很大的。
避免辛辛苦苦注册下来的产品,因为产品名称、适用范围的描述和医院入院的目录名称描述不一致而不能入院销售。严重的情况只能申请注册变更修改产品名称和适用范围的描述,这在时间上的损失是很大的!
3,竞品分析
知己知彼百战不殆,通过竞品调研分析,可以了解该产品的最新行业动态。
在这个时候作为注册工程师,你可以使用国家药品监督管理局的数据库来搜索产品信息,将信息汇总给到项目组,并结合市场部门在市场上调研的结果,与研发部门共同协商。目的是为了给技术部门一些参考,让即将研发的产品更好到找到市场定位,想出自己的产品特点、卖点和优势。
同时还能在审评中心官网搜索到相关的审评报告,可以供我们参考,为接下的注册工作提供有效的帮助。
在整个阶段中注册工程师搜集到的信息最终汇总并输出一份注册可行性调研报告。将注册产品的信息明确在受控文档中,避免在后续工作中产品信息不明确或反复修改。
受控文档整个项目组都可以查阅,会让产品信息同步化,让整个项目组更明确、顺利、有效地开展后续的工作。
在这个阶段,我们注册工程师要具备的是:
检索能力,能够有资源有渠道,更快、更全面地搜索产品相关信息;
沟通能力,能够跟项目团队的各个部门进行有效的沟通,推进项目。
从设计开发的定义我们知道,设计开发是一组过程,有输入和输出。设计开发的输入是与产品有关的信息,也就是项目开发需要哪些条件,按照什么方式进行,时间如何安排,预期效果和预估的风险有哪些?
设计输入过程是一个非常重要的环节!输入为产品的设计和开发提供了基础和依据,决定了产品结构组成、生产工艺、性能评价体系等内容的输出,不仅影响产品研制和生产,在产品全生命周期管理和推动产业高质量发展中同样具有重要作用。设计输入是产品全生命周期的基础,设计输入完成后,就可以开始进行主要研发了。(当然这个时间是灵活的,有些工作可以在各个阶段同时开展)。
总的来说,设计输入是客户需求的反应,代表着对最终产品的要求。就好比我们去买吃的,我们会跟老板说“我要一个,加香肠、加鸡蛋、微辣、酸甜口烤冷面”这句话里我们就给老板输入了我们的需求,老板会按照我们输入给他的需求做(she ji kai fa)一个满足我们要求的烤冷面。
在这一阶段里:
市场的同事会根据市场、顾客等的需求和期望,提出他们的需求——市场需求
技术工程师他会从制造材料、结构设计、工作原理、产品安全、正常使用等方面考虑,总结为产品的需求——产品需求&设计研发计划等文件
工艺工程师的角度来讲,他考虑的是生产工艺相关的信息——制定生产工艺流程、采购生产设备、制定生产计划等规范文件。
质量工程师会分析研究同品种已上市产品的不良事件,从产品的设计开发开始开展风险管理评估,评价产品在安全性、有效性和质量可控性等方面存在的风险。并根据风险的高低采取相应的改进预防措施,确保产品的安全、有效、质量可控。——不良事件分析&风险分析等
那作为注册工程师,应当对法规、标准、指导原则、临床使用等要求进行收集。
在指定产品的性能功能要求和安全要求是,除了考虑用户需求和资深需求以外,最终要的是要考虑法规和标准的要求,做到产品全生命周期的合规性。
这一阶段,注册工程师要输出两份文件《注册临床路径》和《法规标准需求》(可能不同公司有不同的调整和要求,请结合自己公司和项目组的情况适当参考)。
《注册临床路径》是需要我们注册工程师,要制定出整套产品从型式检验到最终获批的过程中的活动和工作,做出明确的计划。
例如:明确产品名称、型号、管理类别、选定检测机构、和最终递交的监管单位,如果产品涉及生物相容性评估、动物实验、临床试验等,需要注册工程师来主导的工作,都要明确出来。相当于注册工作的计划。
《法规标准需求》是相当重要,也是这一阶段注册工程师要着重去整理的内容。这里的法规标准,即指的是法规、标准(通标、专标),还包括药监局发布的各类通知、公告、指导原则等。
如果你所负责的产品预期销售区域不仅仅是中国,那你还需要明确产品预期上市的国家/地区的法规、标准需求和体系要求。
简单的讲,注册工程师需要通过对法规标准的解读,把要求汇总并输入给项目组,这样在产品设计开发阶段就会讲这些需求考虑进去,自然整个产品生命周期是合规的,且生产出来的产品是满足法规标准要求的。
比如,
1,通过对法规标准的解读,可以明确说明书、标签都要包含什么信息、说明书和标签上的标识是否满足法规标准要求(常见问题标签中的内容不满足6号令要求、标识不准确、产品适用的标识未明确在说明书或标签中)。
2,通过对法规标准的解读,可以知道有源医疗器械应用部分的塑料外壳需要满足阻燃等级。设备需要满足防水等级,预期在日本销售的有源医疗设备,电源电压要满足100V等等的要求。
3,通过对标准的解读,可以知道预期与人体接触部分的材料要满足的生物相容性要求,要提前评估并将需求告知采购人员,避免购进的物料不满足生物相容性。物料只能做报废处理,浪费研发资金。
4,不同国家对于生物相容性、加速老化高温高湿的要求,有所不同,提前在预期销售国家的法规标准中找到明确要求,并在设计研发阶段给技术同事提供参考。
像这样的需求还有好多,在设计研发输入阶段多评估出来一些需求,对产品的设计研发出安全有效且合规的产品就会多一些帮助。
当然,同一项目组的其他同事也会在自己的职责里学习和解读与自己职责相关的标准的。
所以,你们看 注册工程师在这一阶段是不是很重要呢。
在这一阶段,注册工程师需要具备的能力:
1,注册临床路径的规划能力,制定合适的规划,选择合适的检验机构和合适的实验方案,是注册工程师必备的能力
2,法规标准学习和解读能力,谁不想成为法规专家呢?
设计输出,顾名思义按照设计输入进入设计与开发的实际阶段,在实际设计开发过程中就会产生输出,在这个设计与开发的实际过程中,如实的记录这个过程的文件、记录,就是我们所说的设计输出记录了。
这一阶段的文件包括:
产品图纸:原材料、零件、组件、半成品、成品图纸、原材料质量标准等
过程文件:工艺开发方案、工艺验证相关活动的计划(无菌内包装、灭菌工艺研究计划等)、IQC、OQC、工艺流程图、工艺规范、生产批记录、设备规范、生产设备清单、生产工装图、操作标准、设备操作规程及维护保养规程等。
物料要求:制定物料接受标准文件
标准测试方法:制定标准测试方法(STM)
产品功能性能:样品试制和测试、设计验证主计划
产品安全性研究计划:生物相容性测试方案、早期动物实验方案(模型)
包装/标签:包装设计、标签设计等
过程检验文件:制定进货、过程、半成品、成品检验规程等
MDR文件:产品技术要求、说明书等技术文件
风险管理:设计故障模式与效应分析、过程故障模式与效应分析等......
在这一阶段中注册工程师要输出注册送检计划、产品技术要求,生物相容性评价方案,等(不同公司情况或有不同,供参考)
注册送检计划要明确:检测中心、检测项目、样品信息、样品数量、包装要求、批次要求、原始记录要求。
产品技术要求:按照《医疗器械产品技术要求编写指导原则》进行编写,需要跟技术部门协商,根据产品的使用目的和预期用途来制定产品性能指标。
生物相容性评价计划:结合GB 16886.1-2022的要求,分析、制定生物相容性评价计划。
除了这三份需要注册工程师主要输出以外,在这一阶段,注册工程师还需要协助其他部门完成其他文件的输出, 比如:协助标签设计(标签内容满足6号令要求)、协助说明书评审等。
设计验证是对产品设计和开发进行验证,来确保设计和开发满足输入要求;设计确认是对产品设计和开发进行确认,确保满足规定的要求和预期用途。
设计验证的定义“通过检查并提供满足特定要求的客观证据进行确认(confirmation by examination and provision of objective evidence that specified requirements have been fulfilled)”(21 CFR 820.3(aa))
设计确认的定义为“通过客观证据确定器械规格符合用户需求和预期用途 (establishing by objective evidence that device specifications conform with user needs and intended use(s) )”(21 CFR 820.3(z))
相信大家都知道,设计验证和设计确认是整个产品生命周期中重要的阶段。在这一阶段,企业应当基于风险评估结果来确定需要进行验证或确认的工作范围和程度,并确保有关操作的关键要素能够得到有效控制。基于风险管理报告中的分析,与验证的一致性。并保存详细的原始数据记录资料。
在这一阶段需要交付:
一,注册检验样品(样品生产批记录,过程检、出厂检记录等)
RA应在设计输出阶段评估样品数量,在此阶段跟进生产进度,确保样品按时交付且注册检测样品生产批记录有详细的保存记录。
二, 注册检验报告
在此阶段,RA要准备在选定的检测机构申请受理检验:
整理编写送检资料(送检资料可以参考之前的推文有源送检-送检送检资料清单);
确认送检样品状态(样品是否经过出厂检、标签是否完整、配件是否齐全);
确认好样品状态后送样,开始测试。
协助检测机构完成测试出具报告。
三,设计验证
设计验证的范围很广,要结合产品本身的功能性能和预期用途并基于产品风险评估结果确定验证活动。主要证明产品的安全性和有效性。
验证活动包括但不限于:
物理特性(尺寸、外观等)、功能/性能、化学/材料表征、包装、货架/运输、稳定性、使用寿命、可用性、生物学评价和动物试验等完整的设计验证等。
预期与其它医疗器械连接后配合使用的产品,应该进行联合使用/兼容性验证;
预期对患者提供能量或物质治疗的产品,需要进行量效关系/能量安全验证;
预期以无菌形式交付,一次性使用的产品需要进行灭菌验证;
预期可处理后重复使用的产品,需要进行满足预期可重复使用次数的再处理验证;
预期需要经过清洗消毒的产品,需要用预期使用清洗消毒液进行清洗消毒验证;
软件和网络安全相关验证等等
看到这里是不是有点熟悉,这些活动的原始数据以及方案和报告就有我们注册递交的资料!也是体系核查必查的资料。
所以在这一阶段,注册工程师除了要完成注册检测、动物试验、生物学试验/评价以外, 还要协助各个部门完成所有设计验证活动。比如:协助解读相关验证测试要求的法规标准、协助确认供应商能力资质、关注各个验证活动的进度,因为他们很可能是导致你不能及时递交注册资料的原因。
为了合理安排注册计划,保证在最后一个验证活动结束,拿到报告后以最快的时间递交注册资料,注册工程师需要制定注册资料中要递交的验证活动和其他资料的checklist,确定验证活动责任人以及预期结束时间,并定期跟负责人确认进度,以确保验证报告和资料的按时交付;
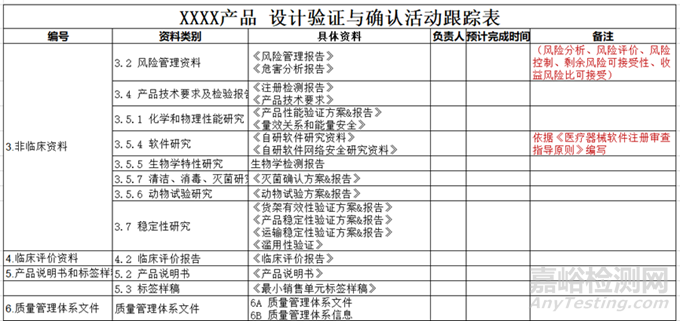
在这一阶段,需要注册工程师具备的能力:
项目管理能力:合理安排时间,跟踪各部门工作进度,确保验证报告和资料按时完成和交付。
沟通能力:跟各部门有效沟通,协助兄弟部门更高效的完成所有验证活动。

来源:医械铁锅炖


