您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-12-12 18:57
一、基本概况
1、自然环境
马来西亚地处东南亚,国土被南海分隔成东、西两部分。西马位于马来半岛南部,北与泰国接壤,南与新加坡隔柔佛海峡相望,东临南海,西濒马六甲海峡。东马位于加里曼丹岛北部,与印尼、菲律宾、文莱相邻。全国海岸线总长4192公里。属热带雨林气候。
2、人口和行政区划
1、人口分布
马来西亚人口约3370万(2023)。其中马来裔70.1%,华裔22.6%,印度裔6.6%,其他种族0.7%。马来语为国语,通用英语,华语使用较广泛。伊斯兰教为国教,其他宗教有佛教、印度教和基督教等。
2、行政区划
马来西亚分为13个州和3个联邦直辖区。13个州包括西马的柔佛、吉打、吉兰丹、马六甲、森美兰、彭亨、槟城、霹雳、玻璃市、雪兰莪、登嘉楼和东马的沙捞越、沙巴。3个联邦直辖区为吉隆坡(Kuala Lumpur)、布特拉再也(布城)(Putrajaya)和纳闽(Labuan)。其中吉隆坡是马来西亚首都,人口约204万,也是马来西亚政治、经济、金融、工业、商业和文化中心。
3、2024年出口概况
2024年1-6月,中国向马来西亚出口医疗器械总计约21.42亿人民币,同比增长约5.21%。马来西亚新增批准医疗器械产品总计2504款,其官方暂不披露制造商国家数据。
二、认证体系
1、马来西亚医疗器械监管机构和法规要求
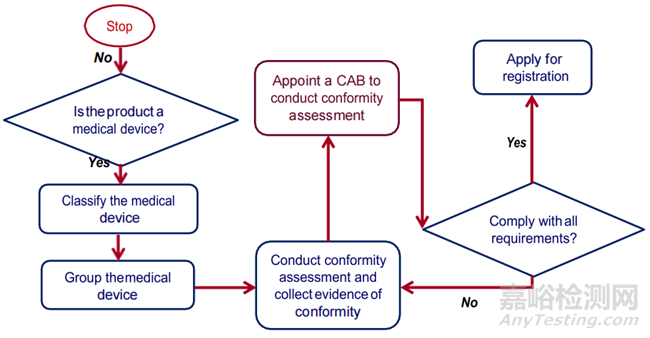
在马来西亚,医疗器械由医疗器械管理局(Medical Device Authority, MDA)进行监管。该监管机构部分监管任务的执行和监督委托了获授权资质的第三方机构(Conformity Assessment Body, CAB)。
监管法规如下:
◆ Medical Device Act 2012,Act 737,即医疗器械法案2012(法案737)
◆ Medical Device Regulation 2012,即医疗器械法规2012
◆ ASEAN Medical Devices Directive 2015,即东盟医疗器械指令
2、医疗器械定义
根据法案737,“医疗器械”指:
(a) 拟单独或组合供人体使用的任何仪器、器具、工具、设备、装置、植入物、体外试剂或校准物、软件、材料或其他类似或相关物品,用于 -
(i)疾病的诊断、预防、监护、治疗或者缓解;
(ii)损伤的诊断、监护、治疗、缓解或者功能补偿;
(iii)生理结构或者生理过程的检验、替代、调节或者支持;
(iv)生命的支持或者维持;
(v)妊娠控制;
(vi)医疗器械消毒;
(vii)通过对来自人体的样本进行检查,为医疗或者诊断目的提供信息,其效用主要通过物理等方式获得,不是通过药理学、免疫学或者代谢的方式获得,或者虽然有这些方式参与但是只起到辅助作用;
(b) 用于人体的任何仪器、装置、工具、设备、器具、植入物、体外试剂或校准物、软件、材料或其他类似或相关物品,部长在考虑公共安全、公共卫生或公共风险问题后,可以通过在公报上公布的命令宣布其为医疗器械。
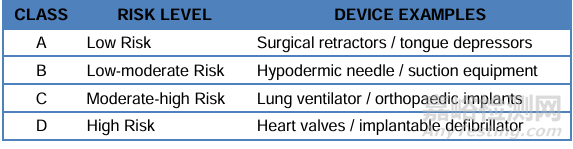
3、产品分类
医疗器械划分为A类(低风险)、B类(中低风险)、C类(中高风险)和D类(最高风险)。
三、注册流程
1、流程
1、General Medical Device
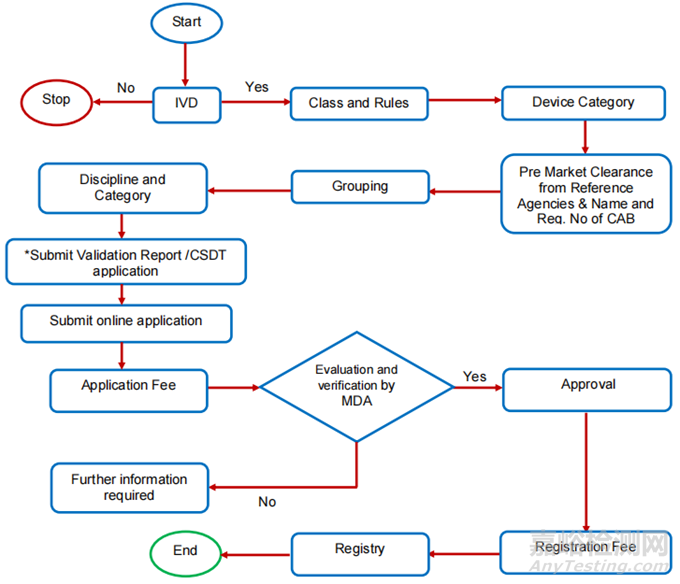
2、IVD
注:Class A(非无菌和无计量功能)不需CAB进行符合性评估;对于无菌及带计量功能的A类CAB仅需审核其无菌及计量功能相关的验证资料。
2、注册申请资料
1、General Medical Device
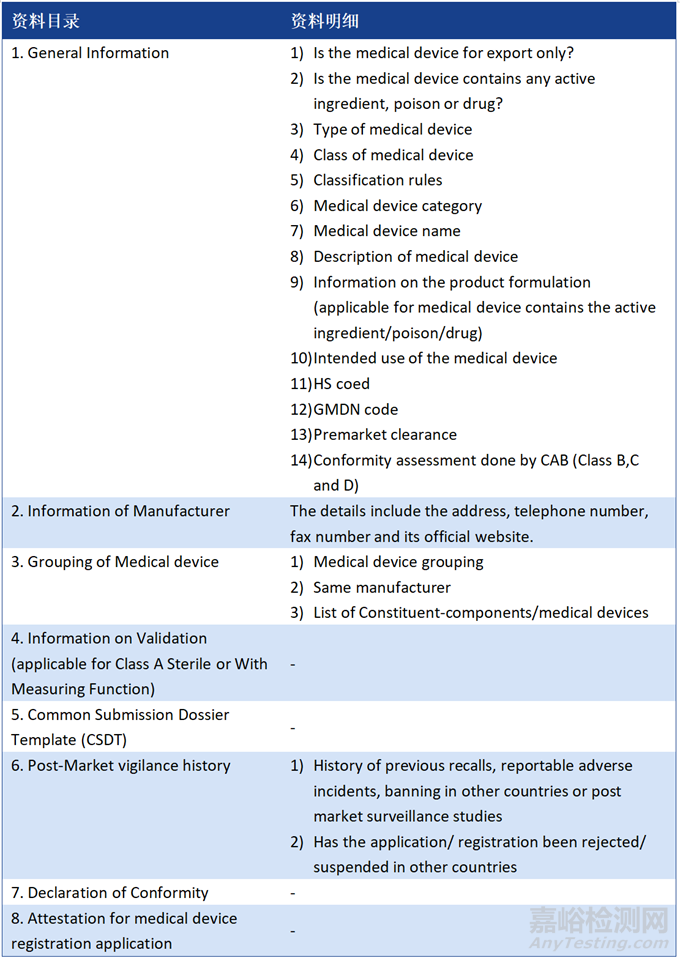
2、IVD
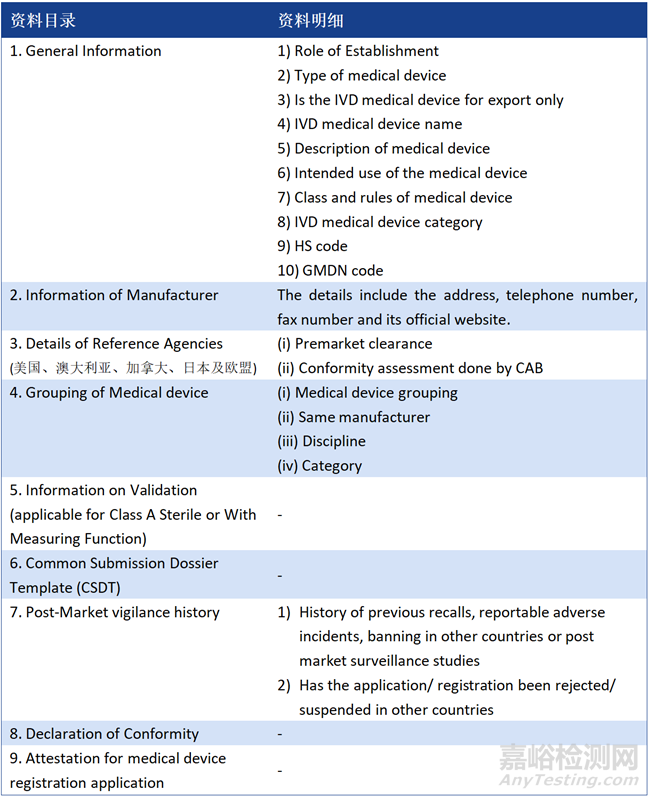
注:资料如有发补,补正时间为90天;家用器械或其他监管机构特殊要求器械的标签及说明书需为马来语(Bahasa Malaysia),除此之外所有文件用英文准备即可,无公证要求。
3、注册周期及费用
1、注册周期:1∼6个月不等
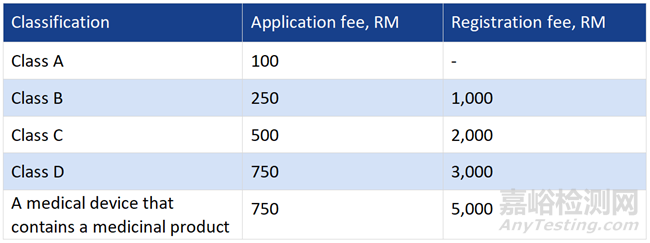
2、注册费用:
4、注册提交
境外制造商需指定一名当地授权代表(Authorized Representative, AR)进行医疗器械注册;授权代表必须是马来西亚公民/永久居民或马来西亚当地的注册公司,并持有场地证书(Establishment License)和医疗器械良好分销规范证书(Good Distribution Practice of Medical Device, GDPMD)。然后由AR通过MeDC@St2.0+系统进行注册申请文件递交。创建MeDC@St2.0+账号及登陆网址链接:https://medcast.mda.gov.my/admin/user/login
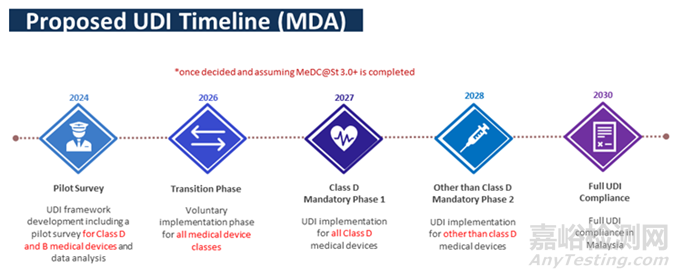
5、UDI要求
马来西亚尚未实施UDI要求,未来计划如下:
6、CAB符合性评估
CAB符合性评估包括以下四个方面的内容:
1)质量管理体系QMS
QMS证书(基于ISO 13485)可由CAB现场审核后颁发;也可以是NANDO公告机构或参考国(美国、加拿大、澳大利亚、日本及欧盟)颁发的QMS证书,但需经CAB审核,是否需现场核实由CAB根据制造商体系情况及产品风险程度决定(一般是少数B类可豁免现场审核)。
2)上市后监督体系PMS
对于B、C、D类产品,CAB需审核制造商上市后监督体系,内容包括:投诉处理Complaint handling, 分销记录distribution records, 强制性器械问题或不良事件上报mandatory problem/adverse event reporting, 现场纠正措施field corrective action, 召回recall。
3)技术文档
采用CSDT (Common Submission Dossier Template),即东盟通用立卷审查技术文档模板。
4)符合性声明Doc
需填写已获批国家(关注美国、加拿大、日本、澳大利亚及欧盟)的医疗器械注册证编号或批准号。
具体详细的符合评估流程、周期及费用等视具体CAB要求而定,符合资格的CAB名单可见链接:https://mdar.mda.gov.my/frontend/web/index.php?r=carian-cab

来源:广东医疗器械学会


