您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-02-17 08:57
导言
医疗器械设计输入必须全面且符合现实需求,要以明确、可验证(能通过客观分析、检查或测试方法)以及可量化的术语来定义。为保证设计输入的充分性,需依据具体产品类型(如体外诊断设备[IVD]、植入式医疗器械、有源医疗设备、无源耗材等)进一步细化输入清单,确保涵盖产品全生命周期管理要求。
此外,由于不同国家和地区的法规要求存在差异,设计输入应按照目标市场的具体法规进行调整,以保证在各个国家和地区的合规性。
一、医疗器械设计输入
1.1设计输入的基本来源
在医疗器械完成设计立项和设计策划后,产品进入设计输入阶段。设计开发立项文件和设计策划文件是产品设计输入的基础内容。具体输入涵盖以下方面:
①用户需求:明确目标用户的期望与使用场景,确保产品具备功能性、易用性和安全性。
②监管需求:满足相关法规标准(如ISO 13485、FDA 21 CFR Part 820、欧盟MDR等)的要求,确保合规性。
③产品需求:定义产品的性能指标、技术规格、材料选择以及制造工艺要求。
④市场需求:结合市场调研结果,确保产品符合目标市场的实际需求和竞争环境。
⑤初始风险管理分析:基于风险评估工具,识别潜在风险并制定相应的缓解措施。
1.2.医疗器械设计输入的规范化要求
①文档化管理:所有设计输入内容均需形成书面记录,并经过评审和批准,确保其完整性和可追溯性。
②多方参与:邀请跨职能团队(如研发、质量、法规、市场、生产等)共同参与设计输入的制定,确保各方面的需求得到充分考虑。
二、医疗器械设计输入法规解读
2.1产品要求的可追溯性
《医疗器械生产质量管理规范》对医疗器械的采购、生产、检验、销售、售后服务有明确的可追溯性要求,虽然在设计开发要求中未明文规定,但也隐含了可追溯要求。GBT 42061-2022设计开发策划部分规定应将“确保设计和开发输出到设计和开发输入的可追溯的方法”形成文件。
2.2明确设计开发输入要求
设计开发输入考虑不全面可能会遗漏部分法规要求。为此,建议充分利用《医疗器械安全和性能的基本原则》,逐条核对其中内容是否适用,若适用,需要考虑的法规标准是什么,需要制定何种指标来满足要求等。对照产品适用的法规标准,检查是否有相关风险遗漏。
2.3充分利用设计开发评审
GBT 42061-2022明确要求设计开发输入应完整、明确,可被验证或确认,且不能自相矛盾。FDA QSR 820也要求,设计开发程序应包括处理不完整、不清楚或相互矛盾要求的方法。GBT 42061-2022和《医疗器械生产质量管理规范》均要求在设计开发策划阶段明确各阶段应进行的评审。如评审认为需修改或更新设计开发输入,可将评审结果合并到设计开发输入中。
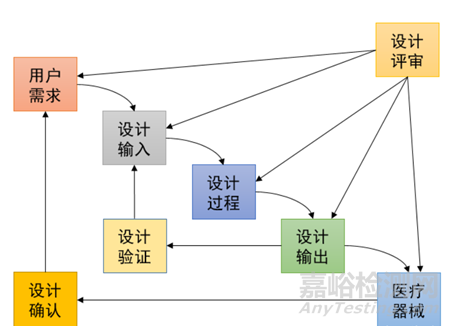
医疗器械瀑布式设计控制过程
2.4持续跟踪法规要求
设计开发输入是一个动态变化的过程,法规标准及规范性文件作为输入的重要组成部分,其要求也会更新。持续跟踪法规要求并分析法规变化对产品符合性的影响,对设计出符合要求的产品至关重要。产品研制过程中有新的标准发布实施时,需评估是否要更新设计开发输入。
三、医疗器械设计输入列表示例
作者是有源医疗器械方面的研发专家,因此对有源医疗器械方面举例。不同医疗器械、不同国家和地区都有不同要求,设计输入都有差异,需根据实际情况调整。

四、医疗器械设计输入输出设计追溯矩阵举例
医疗器械设计输入需设计输入输出设计追溯矩阵,在企业内部正式应用 ,需根据GB/T42061体系中4.2.4文件控制要求编制各项信息,本文由于字数限制,只保留最核心部分。
|
|
|
|
|
|
|
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
|
|
|||||
五、结语
综上,设计开发输入要求是一个不断迭代、逐步优化的过程。很少能一步到位。充分利用设计开发输入评审,形成发现问题、解决问题的良性循环,对解决设计开发输入问题具有重要意义。
本文引用以下两篇文献:
1. 设计开发输入与医疗器械注册申报资料关系探讨 孙克英 陈敏 曹越《中国医疗器械信息》杂志2021年第27卷第23期;
2.医疗器械设计开发控制手册/(美)玛丽·B.特谢拉著;卫根学,潘孔荣,张进进译.-上海:上海世界图书出版公司,ISBN978-7-5192-9150-1;

来源:医械研发笔记


