您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2019-08-15 15:22
近年来,政府在保障公众用械安全和鼓励创新研发方面持续发力,2017年10月,中共中央办公厅、国务院办公厅印发《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,国内医疗器械监管迎来新的机遇和挑战。
本文通过对美国食品药品管理局(FDA)医疗器械上市前通告制度的演变及发展情况的介绍,分析其变化给美国医疗器械产品上市注册带来的影响,为我国医疗器械监管事业提供借鉴。
FDA医疗器械监管的整体情况
美国是世界上最早从法律上对“医疗器械”(Medical Device)作出定义、并对其实行分类管理的国家,其对医疗器械的立法监管已有100多年的历史。为确保医疗器械的安全性和有效性,FDA根据风险等级、上市前安全性和有效性评估需求,将医疗器械分成Ⅰ、Ⅱ、Ⅲ类进行上市前管理。
FDA对医疗器械的上市前准入制度分两种:上市前通告制度(Premarket Notification,即市场预投放登记)和上市前审批申请制度(Premarket Approval Application,PMA)。
上市前通告出自《联邦食品、药品和化妆品法案》第510节第k条,具体规定是:生产企业第一次在美国市场销售产品,或已上市但有重大改变的产品在上市前,应向FDA呈报相关信息,阐明拟上市产品与已上市的参照器械(Predicate Device)在安全性和有效性上是否实质等同。
实质等同(Substantial Equivalence,SE)包含以下含义:第一,拟上市医疗器械与参照器械具有相同的预期用途和相同技术特征;第二,拟上市医疗器械与参照器械具有相同的预期用途,虽不具有相同技术特征,但不会存在安全性和有效性上的新问题。
对于不同医械产品,FDA设定了不同的监管方式。大多数Ⅰ类产品为低风险或无风险产品,如听诊器、压舌板等,仅需进行“普通管理”。对此类产品,FDA豁免上市前通告程序,生产企业只需向FDA提交材料,证明产品符合医疗器械生产质量管理规范,并进行登记后即可上市销售。
对Ⅱ类产品,如计算机断层(CT)扫描仪等,在实行“普通管理”的基础上还需进行诸如附加标签要求等“特殊管理”。除部分产品被豁免上市前通告外,Ⅱ类产品中的多数都需进行这一程序。
Ⅲ类产品一般具有较高的风险或危害性,或属于维持生命的产品,如人工心脏瓣膜、心脏起搏器、人工晶体等。此类产品在上市前需进行临床研究,以评估其安全性和有效性,并需提交上市前审批申请。
上市前通告制度的演进
颁布于1938年的《联邦食品、药品和化妆品法案》首次将医疗器械纳入监管范围。此项法案赋予FDA开展医疗器械监管的权利,但仅限于对假冒伪劣或具有虚假标签的产品提起诉讼。1976年,美国国会通过《医疗器械修正案》,提出对医疗器械实行分类管理,并提出了上市前通告制度与上市前审批申请制度。
至此,FDA对医疗器械的监管框架正式形成。1990年,美国国会通过《医疗器械安全法案》,在《医疗器械修正案》的基础上增加了许多新内容。
首先,《医疗器械安全法案》明确了“实质等同”的定义和判断标准,并提出“参照器械”概念。同时,法案提出“特殊标准”概念,指出:对产品安全有效性的判断除依据检验标准外,还应综合考虑产品上市后表现、患者使用意愿及FDA所认为必要的其他因素,这使技术审评判断标准更加灵活。法案还规定增加上市后监督环节:FDA有权对用于婴幼儿或植入人体等特殊医疗器械开展上市后监督,监督方式不仅包括来自生产企业和监管部门的监督,还包括对不良事件的评估分析、产品召回等方式。
1997年,美国国会通过《食品和药品管理局现代化法》,授权FDA从加速审评的角度进行大范围改革。改革内容包括豁免Ⅰ类医疗器械产品和部分Ⅱ类产品的上市前通告、允许第三方机构开展上市前通告审评、提出对创新产品的重新分类程序等。
FDA规定对全部Ⅰ类医疗器械和部分Ⅱ类医疗器械豁免上市前通告,其判断产品能否受到豁免的标准一般为:产品上市后是否会引发严重的医疗事件、产品设计能否有效避免伤害、产品的各种变更所引起类别调整的可能性等。
同时,为加速审评,FDA允许第三方机构对特定Ⅱ类医疗器械产品进行初审,由FDA根据初审意见作出最终决定。
此外,FDA还设置了重新分类程序。此前,产品获得非实质性等同(NSE)结果后将被自动归入Ⅲ类,需通过临床试验来提供安全及有效性证据。对安全风险不高的创新产品来说,此举会增加生产企业的负担,延缓产品上市。通过运用重新分类程序,获得NSE判断的创新产品可被提出重新分类,归类由FDA确定,归类成功后无须提交上市申请,即可上市销售。
《食品和药品管理局现代化法》还提出了最小负担原则,要求FDA尽可能减轻申请人负担,向申请人提出的申报资料要求为必须出于对产品安全及有效性的验证目的。
此外,2002年通过的《医疗器械用户收费和现代化法》对《联邦食品、药品和化妆品法》相关内容进行了调整,对进行上市前通告程序的审评用户收费。该法案每5年修订一次,对收费标准进行调整。
制度变化对注册审评的促进
缩短审评时长 FDA公布的数据显示:自2002年开始对审评用户收费起,上市前通告的审评按时完成率从77%提高到2008年的93.5%(详见图1)。
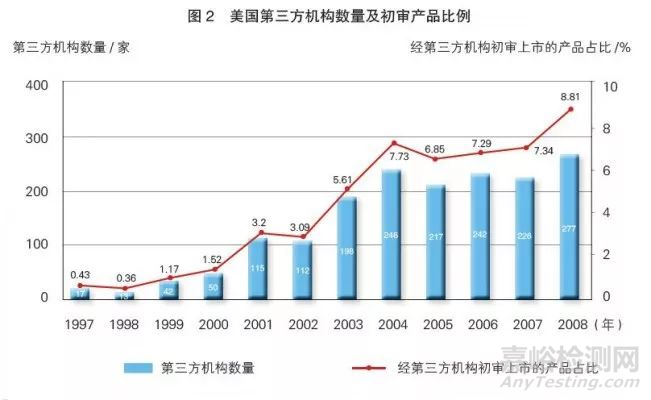
第三方机构的审评项目占比提高 通过对FDA已公布的上市前通告产品进行分析,笔者发现,自1997年美国国会通过《食品和药品管理局现代化法》并允许第三方机构开展上市前通告的初审工作以来,获准上市产品中经第三方机构初审的产品占比不断提高,同时,第三方机构数量亦显著增加(详见图2)。
医疗器械分类编码数量稳中有升 为便于管理,FDA将医疗器械产品按用途进行分类,并赋予分类编码,分类编码的增加量间接反映出新医疗器械产品注册数目的多寡。据FDA公布的数据,自1990年起,分类编码数量稳中有升。截至2009年底,上市前通告产品的分类编码数量已超过2500个,可见FDA鼓励创新的各项措施成效显著。
注册国数量增多 一国监管部门的各项制度可直接影响到境外生产企业在该国的注册申报数量。数据显示,截至2017年,在美上市的境外产品占比超过30%,涉及43个国家和地区。可见,FDA的监管环境得到了世界多国认可。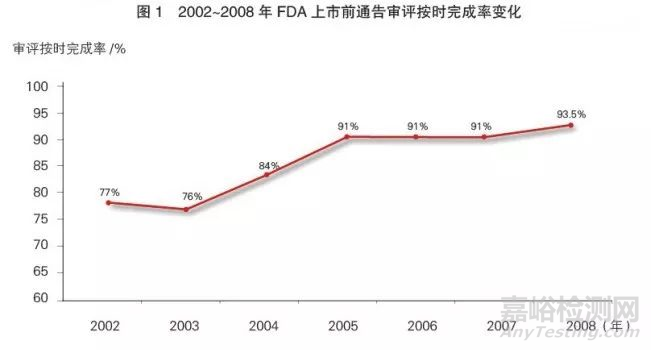
对我国医疗器械监管的启发
美国的医疗器械产业环境面临着应对全球化、鼓励创新等因素的挑战,在此形势下,不断演进、更新的FDA上市前通告制度不仅帮美国医疗器械监管事业成功应对了挑战,还吸引世界各国的生产、研发企业赴美上市。纵观这一制度的发展进程,我们可获得诸多启示:
加快立法,理顺各方关系 上市前通告制度的历次变革均始于相关立法,这令监管部门对制度的实施有法可依。对我国来说,加强医疗器械监管的首要任务是提高法律站位,与之相应,要加快立法工作进程,以此明确国家、省、市各方职责及监管部门同其他部门间的职责分工,理顺各方关系。
提升监管理念,构筑监管科学 FDA公布的监管战略是“推进监管科学的发展与创新”,为实现这一战略目标,其提出“风险-获益理念”“最小负担原则理念”等,并以此为指导制定了各项制度和原则。“战略-理念-实现途径”的监管模式目的性更强,也更加科学严谨。从更宏观的角度制定战略,并围绕战略的实现出台系列规定,将有助于建立符合我国国情与行业特色的监管科学体系。
重视队伍建设,提供智力支持 业务素质过硬的队伍是保证审评工作顺利进行的前提。通过注册申请收费制度等,FDA为改善审评的软、硬件条件(如聘用更多审评人才)提供资金支持。而借助第三方审评机构的力量,也在客观上起到加强队伍建设的作用。
以此为鉴,我国可从以下几方面加强审评队伍建设:增加专业审评人员数量,并根据监管的专业特点合理配置人员岗位;对在岗审评人员加强培训;通过轮岗、借调、挂职锻炼等方式,加强部门及地区间的交流学习;建立合理化激励机制,用制度规范人、激励人。此外,若条件允许,还可鼓励第三方机构承接一部分初审业务,通过合理利用外部审评资源,进一步提高医疗器械的审评审批效率。
来源:中国药事


