您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-03-21 23:23
摘要:随着2017年中国加入国际人用药品注册技术协调会(ICH),中美双报已经成为了本土企业创新药申报的新趋势。首先,本文总结了当前中美双报的现状,截止目前我国已有52家本土企业在中美两国进行新药临床试验申请(IND),但是进行新药上市申请(NDA)的企业相对较少;其次,阐述了中美双报需要考虑的因素,如临床需求、产品研发周期、企业资金、医保政策等;最后,对本土企业中美双报进行策略建议,希望可以为药品成功申报提供支持。
1中美双报的概念及意义
中美双报是指用同一套项目研究资料,同时或分阶段在中美两国进行申报。中美双报可以实现跨国开发,加快药品上市的速度,同时尽量保证中美上市后的产品在质量上一致。由于中美两国对于申报资料的要求不同,需要保证这套资料在药学(Chemical,Manufacture and Control,CMC)、非临床(临床前药理、毒理,Nonclinical)和临床(Clinical)方面同时符合两国的审评标准及技术要求。
在全球化的趋势下,大型跨国药企很少只在一个国家进行一个项目的完整研究,基于全球层面的研究才能真正实现高效开发。随着《总局关于发布化学药品注册分类改革工作方案的公告(2016年51号)》的发布,中国对新药的定义由“中国新”提升到“全球新”[1],实现与国际接轨,间接推动了中美双报的发展。对于制药企业而言,开发新药是一个极其漫长且耗费财力的过程,尽快上市获得利益回报是每个新药研发企业的目标。美国作为全球药品监管水平最高且新药销售额最高的国家,进入美国市场也是中国医药企业的目标。当前,按照FDA的标准研发和申报新药已经成为中国新药开发的新趋势。
FDA PAI检查流程、应对措施与新时代下数据完整性
2017年6月中国加入国际人用药品注册技术协调会(ICH)后,中国的药品研发与注册申报开始与国际接轨。药品审评政策的变化为创新药中美双报创造了积极的条件,一是新的药品注册分类将创新药与改良型新药区分开,引导研发机构和企业明确方向,为我国由仿制药大国到创新药强国的转变奠定了基础;二是进口仿制药和国产仿制药不再进行区分,两者审评标准和质量要求基本统一,有利于中国医药产业融入全球医药产业链和供应链;三是建立了优先审评审批制度,缩短药品审评时间,加快药品上市,对推动创新药研发有重要意义;四是开始接受境外的临床数据,意味着国外已获批的新药无需再做全部临床试验,有利于国外的创新药进入中国市场。
2中美双报的现状及成果
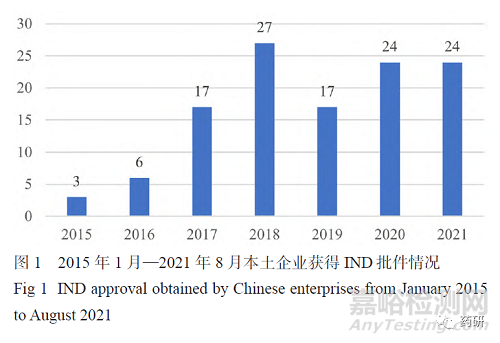
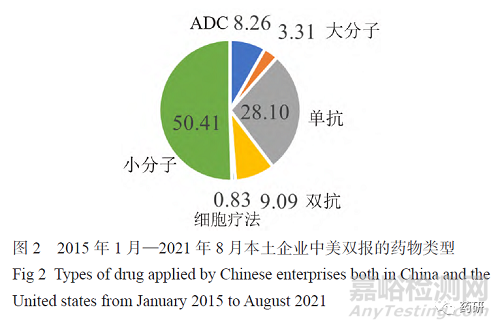
国内最早进行中美双报的企业是恒瑞医药,2009年其向美国食品药品监督管理局(FDA)申请的创新药瑞格列汀获得美国临床试验批件。最近几年,很多国内企业正在积极进行新药中美双报,百济神州、恒瑞医药、亚盛医药、信达生物、荣昌生物等医药企业都获得了一个或多个FDA临床试验批件。根据各公司官方网站、国家药品监督管理局药品审评中心(CDE)临床试验登记信息及 Clinicaltrials.gov 网站披露的信息,本文总结了2015年1月—2021年8月中国本土企业中美双报的情况,涉及中美双报的共有52家医药企业。本次统计按照最后取得中国或美国新药临床试验申请(IND)批件的时间来算,如康方生物AK104于2020年3月取得了美国IND批件,但是2021年1月才获得中国IND批件,本文将其归入2021年获得批件的名单里。图1表明自2017年开始,国内进行新药中美双报的产品数量相较之前几年明显增加。从图2可看出,目前中美双报的产品主要以小分子为主(50.41%),其次是单抗(28.10%),然后是双抗(9.09%)及抗体偶联药物(ADC)(8.26%)。图3列出了中美双报产品的适应证类型,发现绝大多数为抗肿瘤药物。
国内企业的中美双报目前还是主要以 IND 申请为主,新药上市申请(New Drug Application,NDA)相对较少。中国企业第一个完全自主研发在FDA获批的药品是百济神州研发的泽布替尼,该产品于2019年11月15日NDA获 批, 用 于治疗复发性难治性套细胞淋巴瘤。2019年12月20日,石药集团高血压专利药马来酸左旋氨氯地平片(玄宁)成为第二款在FDA获批的中国创新药,该产品是改良型新药,通过FDA505(b)(2)途径申报NDA获批。

3中美双报需要重点考量的因素
3.1 以临床需求为导向
虽然中美双报成为了新的趋势,但并不是所有的产品都适合中美双报。在项目立项前期,进行立项调查时应该充分考察开发产品的临床需求,不仅包括中国市场,还应该考察美国甚至全球市场。由于疾病发病有一定的地域性特点,相同疾病在不同地区的发病率不同,药品的临床需求也会不同。例如,肝癌、食道癌在美国是罕见病,但在中国并不是,那么这两种癌症相关的药物在中美市场的临床需求完全不同。
3.2 产品的研发周期与企业的资金支持也是重点考量因素
目前,我国仍处在由仿制药大国向创新药强国转变的时期,大多数企业的产品还是以仿制药为主,在创新药开发方面缺少经验。一个新药从发现到上市需要很长的时间,且期间还存在失败的概率,这对以仿制药为主的企业来说是巨大的挑战。另外,企业的资金是否雄厚也是需要重点考量的因素,新药从发现到上市需要投入巨额资金,美国仅临床Ⅲ期的费用就高达1亿美元。可见,中美双报对公司的财力、时间投入都有较高的要求,企业在做决定时需慎重考虑。
3.3 其他因素
企业研发团队的能力与新药研发成功的概率密不可分,中美双报还涉及到与 FDA 的沟通,需要海外注册申报的经验,对研发团队在语言层面的要求更高。另外,医保政策、医师的处方习惯也是中美双报需要考虑的因素。
4中美双报策略建议
4.1 利用中美两国的孤儿药政策加速药物研发进度
罕见病指的是发病率极低的疾病,又被称作“孤儿病”,治疗罕见病的药物称为孤儿药。目前中国对于罕见病还没有明确的定义,仅在2018年5月发布了第一批罕见病目录(包括121种疾病);美国将每年患病人数小于20万人的疾病定义为罕见病;日本将罕见病定义为患病人数小于5万的疾病。目前全世界的罕见病超过7000种,绝大多数的罕见病是由于遗传缺陷造成的,发病也具有区域集中性,不同地区发病率不同。因此,针对同一种疾病,在一个国家是罕见病,在另一个国家不一定是,例如胃癌在美国属于罕见病,但在中国并不是。
根据企业官网披露的信息,2020年已经有20多款中国生物医药公司的创新产品获得 FDA孤儿药资格认证,主要包括PD-1/PD-L1抑制剂、CAR-T 疗法、抗体 - 药物偶联物(ADC)、基因疗法等,如君实生物的特瑞普利单抗、康宁杰瑞的KN046、信达生物的信迪利单抗等,这表明孤儿药研发申报已经变成了中国企业进入美国市场的路径之一。
美国于1983年出台了《孤儿药法案》[2],针对孤儿药的开发,给予了一系列的政策优惠,如减免申请费用、降低企业税收、7年市场独占期、有条件批准、专项研发基金资助等政策;另外,FDA在2017年6月发布了《孤儿药现代化计划》[3],规定“对超过120天的所有请求在90天内进行完整性审核,对之后提出的新的孤儿药资格认定在90天内给予回复”,极大地节省了孤儿药的审评时间。目前,中国对于罕见病注册申报在审评时间、税收等方面也给予了政策倾斜。例如,在审评时限方面,新版《药品注册管理办法》规定罕见病药品可以申请优先审评审批程序,对于“临床急需的境外已上市境内未上市的罕见病药物,审评时限为七十日”[4],极大地缩短了审评时间,且允许滚动提交技术资料;在税收优惠方面,国家税务总局规定自2019年3月1日起,进口罕见病药品按照3% 征收进口环节增值税 [5]。可见,中美两国政府都给予了罕见病审评时限及费用等政策优惠,本土企业可以通过罕见病政策加快药物研发及申报进度。
4.2 改良型新药505(b)(2)途径申报策略
1984年,美国《药品价格竞争和专利期修正案(Hatch-Waxman 修正案)》将505(b)(2)作为一种新药申报途径正式予以确定 [6]。中国起步较晚,CDE于2020年发布了《化学药品改良型新药临床试验技术指导原则(征求意见稿)》[7],鼓励中国企业进行化药改良型新药的临床开发。化药在FDA申请主要有505(b)(1)、505(b)(2)和505(j)三种方式。其中,505(b)(1)和505(b)(2)为NDA,505(j)为简略新药申请(Abbreviated New Drug Application,ANDA),适用于仿制药申请。505(b)(1)是全新的创新药申请,对应的是中国化药分类1类新药,在FDA申报时要求申报资料中包含完整的安全性和有效性研究报告,包括CMC、临床前药理毒理研究、药代动力学和生物利用度研究、临床研究等,这些研究必须由申请者开展或者申请者具有使用权。505(b)(2)是指改良型新药,即在已知活性成分的基础上,对其结构、剂型、给药途径、适应证等进行优化,且应具有明显的临床优势、境内外均未上市的药品,对应的是中国化药分类2类,在FDA申报时要求申请者提供完整的安全性和有效性报告,但是报告中的内容并不是全部来自于申请者开展的研究或者申请者无权引用,申请者可从已经公开发表的文献或者FDA审评报告中获得想要的数据。
505(b)(2)申报路径有很多优势,一是它是基于已经批准上市的药物进行的研究开发而提出的申请,有效降低了完全开发一个新药的失败率;二是可利用外部数据进行申报,并且可以通过桥接试验免除部分非临床或者临床试验,减少了相关费用,有效降低了成本;三是美国 FDA给予新药3~7年的市场独占期。
最近几年,中国少数本土企业已经开始探索通过505(b)(2)途径进入美国市场,并且取得了不错的成绩。中国第一个通过505(b)(2)申报途经在美国 FDA 获批的 NDA 产品是石药集团的马来酸左氨氯地平片(玄宁),于 2019年12月19日获批,用于降血压。中国第二个505(b)(2)改良型新药在美国FDA获批的是上海迪赛诺生物医药公司的产品多替拉韦钠拉米夫定替诺福韦片(50 mg/300 mg/300 mg,DLT 片), 于2020年10月6日获批,用于感染人类免疫缺陷病毒1型 (HIV-1)的成人和儿童的临床治疗。还有,绿叶制药 505(b)(2)改良型新药瑞欣妥(Rykindo)于2019年已经在FDA提交了上市申请,还未获批,已经于2021年1月12日获得中国国家药品监督管理局(NMPA)批准上市。可见,通过505(b) (2)申报路径进行NDA逐渐成为中国本土企业中美双报的路径之一,未来还将会有更多的企业通过此途径进行申报。
4.3 利用中美两国审评时间和法规指导原则,寻找最优申报路径
2018年7月11日,CDE发布了《接受药品境外临床试验数据的技术指导原则》[8],宣布可部分接受或者完全接受境外临床试验数据,加快国内临床急需的境外已上市新药进入中国的速度。同时,根据ICH的制度,ICH的成员国也要承认中国的临床数据,为中国本土企业进入欧美市场搭建了桥梁。
企业同时在中美两国进行IND申报还是先后申报也值得考虑,美国FDA对于 IND申请的审评时限是30日(自然日),而中国CDE的审评时限是60个工作日,从时间来看,美国比中国审批得更快一点。另外,从申报资料的关注点来看,FDA更关注体外安全性数据及动物实验数据,CDE不仅关注安全性还关注有效性数据,且有时还会存在资料发补的情况。因此,企业先在FDA进行 IND申请,得到FDA反馈后再在中国申报不失为一种好办法。美国FDA的法规更为完善且原研药审评经验更为丰富,本土企业可以结合FDA的意见再在中国进行申报,提高申报成功率。
中美两国都存在加快审评的程序,中国新版《药品注册管理办法》中列明了以下四条程序:突破性治疗药物程序、附条件批准程序、优先审评审批程序、特别审批程序;美国FDA有快速通道(fast track)、优先审评(priority review)、加速批准(accelerated approval)和突破性疗法认定(breakthrough therapies)四条途径,企业可以充分利用中美双方的这些快速审评途径以加快注册申报。
5 结语
本文阐述了中美双报需要考虑的因素和利用中美双方的法规政策申报的策略,致力于为国内企业中美双报提供建议。但是,在企业申报过程中,还需要结合具体的产品特点、是否存在种族差异、中美法规监督部门对申报资料的要求等情况具体分析,制订更为完善的申报策略,期待越来越多的国内企业中美双报成功。

来源:中南药学


