您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-03-26 02:39
[摘要] 本文对美国非处方药进行研究,发现非处方药具有安全窗宽、无需执业医师指导用药等特点,这些特点决定了非处方药在上市路径、申报资料递交、审评及监管、标签等方面与处方药存在差异。此外,还对美国2020年3月27日实施的非处方药改革进行研究,以期为我国非处方药的申报、审评和监管提供参考。
处方药( Rx) 与非处方药( OTC) 是基于药品自身的安全性、用药过程安全性( 是否需要监测以及用药的复杂性等) 对药品进行分类的一种方法。Rx由执业医师处方用药,OTC不需要凭处方便于消费者购买和使用,对药品实施分类监管,可以合理分配监管资源,也可以大大降低处方成本、就医费用等。
Rx与OTC除了在自身安全性和使用安全性、方便性方面存在区别外,在上市途径、申报资料递交、说明书、包装( 自我药疗会以更小的包装供应)及上市后监管等方面也有所不同。OTC与处方药品的分类不仅与成分有关,还与适应证和药物剂量有关,例如同一活性成分( API) 的不同剂量和/或适应证可能属于不同分类,即所谓“双跨品种”,如高剂量的布洛芬用于治疗关节炎为Rx,低剂量用于治疗头痛及其他疼痛属OTC。
本文就美国OTC的定义和特点、上市路径、申报资料递交、专论程序的改革、OTC审评及监管部门、标签等方面进行研究,以期为我国OTC的申报、审评和监管提供指导。
1、定义和特点
根据美国《联邦食品药品化妆品法》,按照Rx监管的人用药品是指只有在执业医师的指导下使用才能保证安全的药品,包括毒性较大或具有其他潜在有害反应、使用方法复杂或使用时需要监测的药品[1]。
OTC药品应当具有以下特点: 安全、有效且安全性范围宽,作为OTC药品获益大于风险; 误用和滥用可能性较低; 患者对需治疗的疾病可以自行诊断; 不需要执业医师进行安全、使用方面的指导; 标签内容足以使消费者能够自我诊断、自己选择、自行服用、决定何时停药[2]。
在进行OTC的风险和获益评价时,FDA通常会考虑消费者是否可以理解并遵守标签、说明书的内容进行服药,患者是否可以自行诊断疾病,或至少了解其拟治疗的症状,以及在持续使用药品的过程中是否需要进行体检或实验室检查[3]。
2、上市路径
OTC主要可划分为2条上市路径,即注册路径( NDA、ANDA或补充申请) 和专论路径( 修订专论或按专论申请上市) ,见图1。
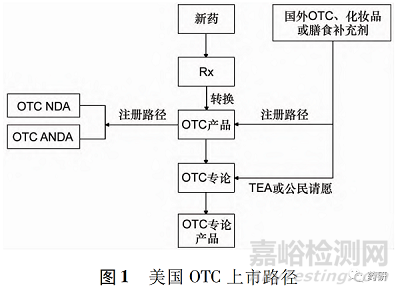
2.1 注册路径
2.1.1 新药申请或者仿制药申请路径 新化学成分、新的复方组成或已有化学成分的新适应证、新剂型或新给药途径,通常情况下按新药申请(NDA)。
企业须按照新药申请的格式递交相关数据,以证明产品作为OTC预期用途的安全性、有效性。按照新药路径批准的OTC,其数据保护期一般按NDA药品的保护期执行。也可以以仿制药形式,按照仿制药申请( ANDA) 路径上市。
2.1.2 已有产品的 Rx-OTC 转换( 补充申请或新申请) Rx-OTC转换是指先前按照Rx上市的产品以相同的适应证、规格、剂量、使用方法、使用疗程、剂 型、使用人群和给药途径按照非处方上市( 完全转换) ; 或按照Rx上市的药品,其活性成分仍然属于Rx成分,但上述其余内容被拟定为OTC产品( 部分转换) [4]。
无菌制剂关键技术解析与发补情况、现场核查
FDA PAI检查流程、应对措施与新时代下数据完整性
清洁验证、工艺验证、设备确认及验证主计划
M4格式申报资料撰写与药品注册现场核查要点解析
考虑Rx 在转换为OTC之前通常已经上市多年,Rx-OTC转换受到内在特殊不良事件和外在自我选择理解能力、使用说明、自我给药等因素的影响。Rx-OTC转换无剂型限制,凡是批准的剂型都可以转换[2]。提出转换申请的主体可以是批准Rx的上市许可持有人或其他主体。
大多数Rx-OTC转换都属于部分转换,即活性成分仍然属于Rx,而特定的适应证、规格或剂型通过NDA申请获批OTC,例如,局部抗真菌剂治疗足癣、癣菌病和股癣时,属于OTC药品,而治疗花斑病时,属于Rx[5]; 另一种情况是Rx-OTC完全转换,通过新药补充申请( sNDA) 转换,活性成分不再属于Rx成分。例如MiralaxⓇ ( 聚乙二醇) 、RhinocortⓇ 抗过敏喷剂( 布地奈德) 等[6]。
截至2016年5月4日,美国约有40种Rx的活性成分通过转换成为OTC的活性成分[3]。1980年之前都是通过专论审核途径转换为OTC,大部分是批准活性成分转换,而不对单个产品进行转换。转换过程中FDA考虑的关键问题是患者自己使用药品时,是否可以达到预期的疗效而不产生安全性问题。转换的重点研究工作是证实标签的可读性、能否自我选择( self-selection and deselection) 、依从性等。这就要求转换后的OTC说明书内容须做到通俗易懂,突出重点信息[7]。如果拟定增加新适应证或新患者人群时,还需要进行消费者研究以评价产品成为OTC的可能性,原Rx 标签中需要解决的问题以及产品的临床使用。OTC转换过程中需补充临床试验数据的,一般给予3年的市场独占期作为激励[8]。
2.2 专论路径
自1972年建立专论以来,目前,美国列出的OTC专论涵盖90多个治疗类别,800多种活性成分,1400多种适应证[9 - 10]。
OTC专论明确了上市OTC活性成分的安全性、有效性及标签内容要求。OTC专论持续更新,根据需要增加新的活性成分和标签内容。专论的修订可以采取时间和范围覆盖申请( time and extent appli- cation,TEA) 程序和公民请愿程序。
2.2.1 TEA 程序 所谓TEA程序是指尚未在美国以药品身份上市的产品纳入OTC专论体系,需要提供在其他国家或地区作为OTC 药品上市使用的历史,至少在一个国家连续销售5年以上,并有足够的使用人群。FDA基于资料评价TEA申请是否可以直接纳入OTC专论。目前已有多个药品按照这种途径获得FDA批准[11]。
FDA法规对纳入OTC专论的药品建立了标准和程序。通过TEA程序纳入OTC 专论可以是以特定“条件 ( condition) ”纳入( “条件”包括: 活性成分或植物药成分的描述、药理类别、拟定OTC用途等) ,也可以要求创建新专论。在 TEA框架下,美国以外按照化妆品或膳食补充剂上市的产品可以以OTC 药品在美国上市。如果OTC药品的“条件”仅在美国以外的一个国家获得批准或者在1972年开始的美国OTC药品审评以后,那么该产品只能按照联邦食品、药品和化妆品法( FD&CA) 505条款通过注册途径获得上市批准。
FDA通常在收到TEA申请的1年内做出是否有资格纳入专论的决定。对于纳入最终专论的“条件”,法规要求活性成分或植物药成分需收载于美国药典专论,并阐明其鉴定、规格、质量和纯度等标准。
2.2.2 公民请愿程序 通过公民请愿的方式可以申请修订或撤销拟定或最终OTC专论中的“条件”。公民请愿程序不可以用于TEA相关申请。公民请愿程序公开、不收费。以往通过提交公民请愿程序修订OTC药品专论通常比新的NDA申请用时更长。
2.3 专论修订程序的改革
2020年3月前,OTC专论的增加、删除或修订需要经过拟议规则制定的提前通知、暂定最终专论和最终专论等3个法定程序[12]。这种采用法规程序增减OTC专论的方式,在解决重大安全性问题时,无法及时对 OTC 专论进行修订。2020年3月27日美国通过了《新冠病毒援助纾困经济安全法》 ( Coronavirus Aid,Relief,and Economic Security Act,简称CARES 法案) ,该法案中倡导对美国OTC专论药品的监管进行改革和现代化,旨在通过行政命令程序代替法规程序对专论增加、删减或修订内容,行政命令程序有助于在OTC药品审评过程中提高效率、提升时效性和增强可预测性,从而促进创新,建立快速解决安全性问题的机制,尽早完成待定专论的审评[13 - 14]。企业和FDA均可发起行政命令程序,企业递交OTC专论命令申请( OTC monography order request,OMOR) ,FDA负责受理、审评、拟定命令、发布最终OTC专论。当药品出现紧迫的安全性问题或更改药品标签、药品分类等缓解与药品使用相关的重大或严重风险时,FDA可以使用加速程序发起命令。
随着OTC专论的改革,某些按照OMOR申请对OTC专论最终命令进行变更的申请人可能获得18个月的市场独占期。包括药品中含有的活性成分( 活性成分的酯基或盐基) 尚未包含在非NDA申请的某些非处方药品中; 或对药品使用条件进行变更,而申请人为变更使用条件所开展或申办的新的人用数据研究对命令的颁布起了关键作用。OTC专论的其他变更不会授予市场独占权,例如安全相关变更,或其他FDA认为是为确保安全使用所做的必要变更、安全性或有效性检验方法的相关变更等。
2.4 按专论申请
OTC专论按治疗种类分类,每个治疗种类包括多种活性成分; 同一活性成分也可能被列入多种不同类别的OTC专论中[10]。如果OTC符合专论,企业在制定成分表/标签时可参考专论,无需 FDA 批准即可上市销售,若不符合专论,则需进行审评[15]。
由OTC活性成分组成的复方制剂,除了可用于单个成分治疗适应证的安全性和有效性之外,OTC审评程序还为某些被认为是安全有效( GRASE) 的成分的固定组合产品上市提供了依据。
3、注册路径与按专论申请路径的比较
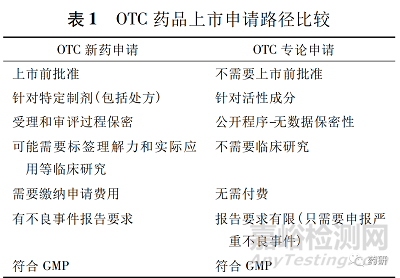
申请人可以按照505FD&CA注册路径进行新OTC药品“条件”的申请或申请变更最终专论的OTC药品“条件”( 21CFR330.11) 。按照注册路径( 505) 获得特定产品的批准与公民请愿程序TEA程序相比,有一定的优势。例如,批准过程保密,某些产品可能获得一定的市场独占期,批准所需时间更短。但注册路径( 505) 批准也有其劣势: 仅针对特定药品( 包括剂型和标签) 的批准; 除了不良事件报告外,还有其他报告要求; 某些药品后续变更标签和剂型需要提交变更申请,这些申请可能需要缴费。
新药、仿制药或补充申请和专论审评都是 FDA基于科学审查做出的决策,见表 1。
4、OTC 审评及监管部门
美国的OTC的审评和上市后监管均在CDER。伴随着2020年3月27日OTC改革,FDA对OTC的组织机构也进行了改革[16]。原OTC的审评、监管是在 CDER新药办公室下设的OTC处( Division of Non- prescription Drug Products,DNDP) ,改革后则由新成立的OTC办公室( Office of Non-Prescription Drug, ONPD) 负责OTC的开发、审评及监管[17]。OTC 办公室又分为OTC Ⅰ处和Ⅱ处( DNDP Ⅰ和Ⅱ) ,每个处负责不同OTC适应证。
OTC处负责OTC的INDs,NDAs,sNDA 的注册审评及上市后监管包括Rx-OTC转换。OTC审评时,OTC处是领导部门,负责组织、协调其他办公室提出的有关科学建议的审评( 合作或咨询) 、召开审评会议等。新药办公室的多个部门会参与OTC审 评,例如有效性数据可能需要Rx 审评部门的医学/统计学专业意见,致癌性或其他动物毒性数据可能需要药理毒理专业提供意见等。除了OTC处,另一个参与OTC审评的重要部门是专题评审处( Specific Subject Matter Review Division,SSMRD) ,专题评审处属于新药办公室下设的各审评处,负责按生理疾病分类的临床资料审评( 例如心血管药物) ,OTC处和专题评审处分工明确( 见表 2) 。在 NDA阶段,OTC处负责审评消费者研究资料、上市后安全性数据、 OTC标签以及所有法规监管问题,在该阶段,专题评审处与OTC处一起对临床试验相关的有效性、安全性数据进行审评。必要时,也可以咨询临床药理学、统计学及化学部门意见[18]。

非处方药咨询委员会 ( Nonprescription Drugs Advisory Committee,NDAC) 协助OTC办公室评价OTC专 论 产 品,目前已有30万种OTC药 品 上市[19 - 20]。CDER 合规办公室的OTC药品组负责消费者用药风险管理。
5、OTC 标签
由于OTC无需医生处方,无需医师或药师指导和监督用药,故其说明书是消费者了解药品的主要途径,消费者对药品的认知直接影响了用药的正确性。
OTC标签必须包括预期用途、说明和警告,以便阅读理解能力低下者都能理解。先前有研究发现美国OTC标签的可读性较差,难以找到产品说明、警告、批准使用及过敏信息。在美国,30% 的OTC由老年人购买,他们认为适应证、慎用、禁用等词不容易理解[21 - 22]。
为提高OTC标签可读性,FDA于1999年制定了OTC 标签可读性法规,规定使用消费者友好型语言及进行格式标准化,使标签的语言更容易理解,消费者知晓重要的信息所在位置。2010年,FDA出台了《非处方药标签理解力研究指南》,FDA认为随着人们教育程度和自我保健意识的不断增强,有效地传递用药信息将成为当务之急。
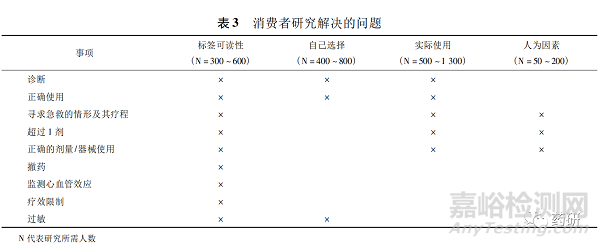
为了了解消费者对说明书内容的理解程度, OTC需要进行特殊的消费者研究[23]。消费者研究类型有标签可读性研究[24]( 对标签关键信息的理解力) 、自己选择研究( 选择正确的产品) 、实际使用研究( 按照标签上的说明使用) 、人为因素研究( 药物相互作用) ,见表 3[2]。
截至2021年6月28日,FDA标签数据库收载人用OTC药品标签有90707 个,人用Rx 和生物制品标签50485 个[25]。
6、讨论与建议
6.1 根据OTC的特点,建立简化的OTC审评路径,区别于Rx审评路径
美国OTC上市路径主要包括专论、Rx 转换、直接申报OTC以及成熟OTC改良等。从上市监管部门来说,美国OTC药品上市前的审评及上市后监管都是由CDER负责,我国上市前审评、审批由药品审评中心负责,Rx-OTC转换由药品评价中心负责; 从上市路径看,我国未设立OTC专论路径; 从注册申报路径看,我国OTC与Rx 从审评资料递交、程序、路径、上市后监管等方面无差别[27]。我国从2000年开始实施Rx和OTC分类管理,新修订的《药品注册管理办法》明确了Rx和OTC实行分类注册和转换管理[28]。分别由药品审评中心和药品评价中心制定OTC上市注册相关和上市后转换相关技术指导原则和程序。药品审评中心于2020年7月6日发布了《化学药品非处方药上市注册技术指导原则( 征求意见稿) 》[29]。
6.2 建立患者友好的 OTC 说明书规则
对于药品说明书,我国有OTC范本,但说明书范本的内容与Rx 的说明书无差异,其表述方式晦涩难懂,存在可读性差的问题,尤其是中药,存在诸多尚不明确的内容,这不利于患者更好地理解说明书内容,可能造成用药安全和有效性问题,应加强对OTC的上市后研究和监测,及时修改药品说明书,更好地指导公众合理用药。目前,我国说明书理解力研究和监管要求仍属空白[30],这也导致了我国OTC药品说明书管理中存在诸多问题[31]。说明书内容能否很好地被消费者理解是保证消费者安全、有效使用OTC的关键,故建议我国参照美国开展消费者研究,并建立OTC标签理解力研究和自己选择研究等指导原则,指导申请人开展相关研究。
除了说明书外,提供更多途径使消费者了解OTC药品信息,加强培训和广告监督,提升自我药疗能力,避免在用药决策上产生误区,出现不合理用药和滥用药物等情况。双跨品种从包装设计上进行区分,避免消费者用药混淆,将消费者用药安全风险降到最低。
6.3Rx 转换为OTC时,若需要额外的临床研究,应适当给予市场独占期鼓励
对于潜在适用于作为OTC的药品,鼓励OTC的申报和及时转换,对于按照创新药申报的OTC或需要做临床研究支持Rx转换为OTC的情况,应当给予适当的市场独占期激励。利用现代技术允许更多的药品以OTC身份上市,增加 OTC药品数量,节省监管资源、医师资源,更好地利用资源。

来源:Internet


