摘 要 / Abstract
目的:通过介绍510(k)(上市前通知)第三方审查计划及优化措施,为我国医疗器械审评制度创新提供相关参考。方法:查阅FDA官网相关指南和文件,对第三方审查机构认可资质、审评程序、监督机制进行介绍。结果:第三方审查计划的实施对弥补监管资源、缓解审评压力有所帮助。结论:建议我国可以尝试引入第三方机构协助承担部分医疗器械上市前技术审评任务,减轻监管部门工作负担,提高审评效率和质量。同时需要规范审评流程、完善相关配套法律法规、重视沟通交流机制的建立及加强对第三方机构的监督。
Objective: To provide insights for innovating the medical device review system in China by introducing the 510(k) (Premarket Notification) Third Party Review Program of the US FDA. Method: By studying relevant guidelines and documents on the FDA website, an introduction is given on the accreditation qualifications of third-party review organizations, review procedures and the oversight mechanism. Results: Implementation of the third party review program is helpful to supplement regulatory resources and ease review pressure. Conclusion: Third party organizations may be introduced in China to assist in the pre-market technical review tasks of some medical devices, with the aim of reducing regulatory burden and improving review efficiency and quality. At the same time,efforts should be made to standardize the review process,improve relevant laws and regulations, facilitate communication mechanisms, and strengthen supervision of third-party organizations.
关 键 词 / Key words
510(k);第三方审查计划;医疗器械审评制度
510(k); third party review program; medical device review system
1、引 言
与药品相比,医疗器械门类更多,学科跨度更大。两者都是关系国计民生和人民生命健康的特殊商品,必须严格做到安全、有效与质量稳定。产品的质量体系及其运作水平是影响产品质量的重要因素。随着科学技术的迅猛发展,尤其是计算机和人工智能的快速发展,医疗器械产业随之高速发展,其产品与质量体系越来越复杂,药品监管部门对纷繁复杂的医疗器械产品与产品质量管控体系进行审计和检查的工作量和难度与日俱增。国务院于2015年发布的《关于改革药品医疗器械审评审批制度的意见》提倡:“将食品药品监管总局列为政府购买服务的试点单位,通过政府购买服务委托符合条件的审评机构、高校和科研机构参与医疗器械和仿制药技术审评、临床试验审评、药物安全性评价等技术性审评工作。”[1]2017年10月,中共中央办公厅、国务院办公厅印发的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》提倡:“将药品医疗器械审评纳入政府购买服务范围,提供规范高效审评服务。”[2]在美国、欧盟等发达国家和地区,第三方机构已广泛参与医疗器械产品的审评和质量管理体系核查等活动[3],社会力量得到充分利用,监管效率也逐步提升。通过研究和学习美国关于第三方机构参与医疗器械产品的审评和质量管理体系核查的具体做法,对于我国深化医疗器械行业发展具有重要意义。
2、510(k)第三方审查计划概述
510(k),又称为上市前通知,是产品进入美国市场的一种上市途径。任何国家的设备销售方计划在美国开展销售都必须先向美国食品药品监督管理局(FDA)提交上市前文件,证明自己即将上市的设备与市面上已经合法销售的设备具有同等安全性和有效性[4],且只有通过了510(k)审查,设备才可正常上市销售。《联邦食品药品和化妆品法案》(Federal Food,Drug and Cosmetic Act,FD&C Act)第523条规定,允许经过认可的第三方机构审查某些中低风险的医疗器械。FDA并于2020年3月12日颁布了《针对行业、食品和药品管理局工作人员和第三方审查组织的第三方审查方案指南》[510(k) Third Party Review Program Guidance for Industry Food and Drug Administration Staff and Third Party Review Organizations][5],该指南对第三方机构的准入资格、审评的设备范围及审查的具体程序等都进行了严格规定。美国实施510(k)第三方审查计划,旨在帮助FDA更快地做出审评决策,为医疗器械制造商提供自愿的替代审查程序,使FDA可将资源和精力主要集中在高风险设备的审查上,但同时掌握第三方机构所审查的低风险设备的最终决定权。
2.1 第三方机构认可要求
第三方机构的认可包括首次认可和继续认可。FDA法规文件主要从政务信息、防止利益冲突、人员资质及保证声明四个方面作为首次认可的考虑因素。第三方机构需要向FDA提供基础信息,同时保证参与510(k)审评的人员与设备销售方之间无利益关联,防止任何可能影响审查过程的利益冲突出现,并签署保证声明。
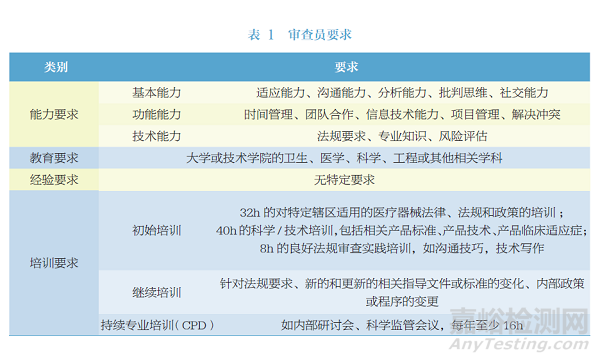
其中FDA对于审评人员的要求采用的是国际医疗器械监管者论坛(International Medical Device Regulators Forum,IMDRF)2017年发布的《监管审查员的能力、培训和行为要求》(Competence,Training,and Conduct Requirements for Regulatory Reviewers)[6](文件编号170316),具体要求详见表1。
2.2 第三方机构审评程序
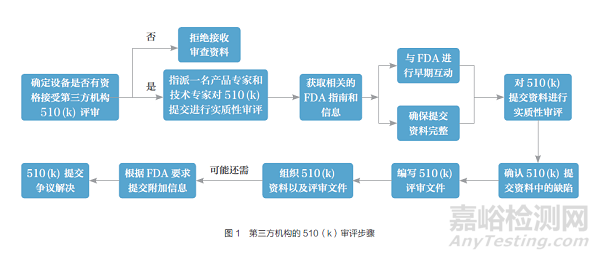
第三方机构具体审评程序主要分为7步,具体流程如图1所示。
2.2.1 确定第三方机构审评范围
FDA根据医疗器械风险高低、特殊控制或一般控制是否足以对医疗器械的安全有效性提供合理的保证等因素将医疗器械划分为Ⅰ类、Ⅱ类和Ⅲ类,只有风险较低的I类及部分Ⅱ类医疗器械能够参与第三方审评计划。第三方机构在开展审评前需要根据产品代码分类数据库或FDA第三方审评公共网站进行设备审评资格的查询,同时还需判断自己是否具有审评该设备类型的专业知识和能力。只有两个条件同时满足,才能开展审评工作。FDA审查部门表明,第三方机构提交的低风险设备的510(k)审评意见不需要复审,而复杂设备的510(k)重新审评可能性较大。为了减少复审次数,提高审评效率,FDA计划调整审评名单,将需要FDA额外审评的医疗器械从名单中移除。
2.2.2 确保提交资料的完整性
第三方机构根据FDA相关法律法规要求对申请者提交的510(k)进行验收评审,查看资料的完整性。如果资料不完整,第三方机构应拒绝开展审评。
2.2.3 实质性审评
实质性审评的重点是确定申请上市的医疗器械是否与市面上已经合法销售的医疗器械有着相同的安全性与有效性。第三方机构需要大量查阅FDA的相关数据库,获取有关的指导文件及信息,如上市前评审信息、上市后召回、医疗器械报告,以及申请者提供的书面反馈、会议纪要及其他数据。FDA 也会为第三方机构提供培训和专家咨询等帮助其完成审评。
2.2.4 早期互动
510(k)审评期间,第三方机构会与FDA进行早期互动,尤其是在审评从未接触过的医疗器械类型之前,这有助于第三方机构了解FDA对该设备类型最新的相关指导原则、标准和其他考虑事项,识别相关问题,确保审评内容和现行审评标准一致。沟通方式通常采用电话或邮件。
2.2.5 缺陷处理
第三方机构在审评过程中发现任何缺陷应立即联系510(k)提交人。第三方机构可以采用电话、传真、电子邮件或信函等方式来解决问题,避免仅通过电话交换实质性数据和信息,需要有书面请求和答复,同时必须进行保密和记录。如果510(k)申请者为了解决缺陷对提交资料进行了任何修改,第三方机构需要进行实时记录,并要求510(k)申请者及时提供510(k)的最新版本。
2.2.6 编写审评文件
第三方机构在提出审评建议时需要编写审评备忘录,提供定制的审评备忘录模板,说明做出最终建议的理由和步骤,并且清楚地记录所有缺陷、对缺陷的响应及对审评的响应。FDA为第三方提供了定制的审评备忘录模板。审评备忘录是FDA对第三方机构审评工作情况进行了解的唯一途径,清晰简明的文件可减少FDA对510(k)的审评次数,提高审评效率。
2.2.7 提交510(k)资料和审评意见
最终审评人员在完成510(k)审评意见的审核后,应向FDA文件控制中心提交两个独立的文件,即申请者提交的510(k)提交文件(最新版本)和机构编写的审评文件,其中审评文件一般包括附有最终审查员签字的相关基础信息表、申请者授权给第三方机构的510(k)、保证性声明、文档目录、早期交互信息、审评备忘录等。
FDA只有在必要文件齐全后,才会对第三方机构的审评备忘录进行审评,同时可能也会重新审评申请者提交的510(k)。如果FDA认为需要额外的信息,将发送电子邮件通知第三方机构暂停工作,并要求其提供附加信息。
2.3 监管机构对第三方机构的监督
第三方机构在通过首次认可之后还需要接受FDA的继续认可,继续认可由医疗器械和放射健康中心(Center for Devices and Radiological Health,CDRH)的投诉监督办公室(OC)依法定期开展(至少每三年一次),检验第三方机构是否按照法规的要求进行相关审核工作并记录过程,并且依然满足认可标准。继续认可因素包括第三方机构过去上市前的评估表现和正在进行的定期评估表现。良好的历史记录和现行审查业绩记录可表明第三方机构能够出具与FDA等效的建议和评审。FDA通过使用公开可用的指标(第三方审评组织绩效报告)来监控第三方机构和整个审评计划的实施,定期进行持续改进分析,以确定计划中可能需要改进的部分,并进行相应地调整[7]。
当有足够的证据表明,第三方审核机构与申报者之间存在着利益关系,或未按照标准和法规的要求履行程序,对公众健康造成威胁时,FDA可以在提供非正式听证会的机会之后采取措施暂停或撤销第三方审评机构资格。
3、对我国医疗器械审评制度的思考
3.1 引入第三方审核的优势和局限性
3.1.1 优势
虽然我国形成了以国家药品监督管理局医疗器械技术审评中心、省级医疗器械审评中心及2个医疗器械技术审评检查分中心(国家药品监督管理局医疗器械技术审评检查长三角分中心、国家药品监督管理局医疗器械技术审评检查大湾区分中心)构成的医疗器械技术审评体系,并通过招聘、编制、借调等方式不断引入人才、扩充技术审评力量,但是审评资源相对缺乏的现状仍然存在,审评人员的数量与高质量完成审评任务的需求还存在差距。引入第三方机构参与审评工作,可以有效缓解监管部门审评资源不足的情况。以美国统计的平均值为例,监管部门对风险较低的简单医疗器械进行单独审评需要105天,在第三方机构辅助审评需要74天,缩短了31天;同样,对风险较高的复杂医疗器械单独审评需要156天,在第三方机构辅助为83天,缩短了73天[8],审评效率显著提升。
3.1.2 局限性
虽然引入第三方机构力量可以获益,但是监管部门若无法对第三方机构进行有效地监管,则该措施亦存在一定的风险。一方面由于第三方机构专业技术能力不足或者不同审评员对法规要求的理解不同,对产品获益-风险评估的标准不同,主观性较强,容易导致审评结果出现纰漏,使得安全有效性存在较大风险的问题产品上市,造成安全事故发生的风险。例如2011年法国某公司的植入性产品通过第三方机构完成CE认证后上市,引发了多例不良事件,之后法国的卫生部门重新评估后认为该产品对人体存在不可接受的风险,不宜上市。另一方面是信任风险,第三方机构作为商业组织,存在一定的逐利性,可能难以避免地会出现权力寻租的现象,一旦发生,将给人民群众的安全带来巨大风险。因此,如何对第三方机构进行监督和管理是该计划实施过程的重点难点之一。
3.2 其他国家或地区引入第三方机构情况
3.2.1 欧盟
欧盟的第三方认证机构称为公告机构,是由欧盟各成员国指定并通过所在成员国相关认证机构的认可后成立的。欧盟委员会将评估合格的公告机构名单公布于NANDO(New Approach Notified and Designated Organisations)网站[9],设备制造商可通过浏览该网站,自行选择公告机构进行医疗器械合格评定。合格评定结束后,公告机构会为其颁发CE证书,该医疗器械才可在欧盟市场中流通和使用。与美国不同的是,欧盟公告机构不仅拥有产品技术资料审评权,还能直接决定产品能否上市并负责产品的上市后监管。欧盟目前采用最新版医疗器械法规[Regulation(EU)2017/745,简称MDR]对公告机构进行严格管理。
3.2.2 加拿大
加拿大将医疗器械分成四类,其中I类风险最低,IV风险最高。医疗器械上市前审评实行的是注册资料审查和质量体系审查相结合的方式[10],与我国相类似,但其质量体系核查是由加拿大医疗器械符合性评估系统(CMDCAS)认可的第三方认证机构(Canadian Medical Devices Conformity,CMDCAS)承担,指导文件为GD210∶ISO 13485∶2003质量管理体系审核[11]。加拿大卫生部仅接受第三方审核机构颁发的质量体系证书。Ⅱ、Ⅲ、Ⅳ类医疗器械在申请注册时,需要提交第三方机构出具的医疗器械质量管理体系认证证书。
3.3 启示
3.3.1 审评机制探索,开展试点工作
鉴于美国和其他发达国家和地区第三方审核计划所取得的良好效果与经验,笔者建议我国可尝试开展第三方机构辅助审核试点工作。美国的第三方审核计划遵循自愿原则,由申请人将费用直接支付第三方机构,FDA不收取任何费用,使得第三方机构可以获得适当利润,也可缩短设备的审核时间,加快上市速度。国外现在已经有许多较为成熟的第三方审评机构,如英国标准协会(British Standards Institution)等。我国可以先与跨国认证机构进行合作,实现对人才、先进技术、理念和审评标准的引进[12],选取少数地区开展试点工作,并确定审评目录,可将第一类备案产品和少部分风险较低的第二类产品交与第三方审评,但第三方只负责给出其审评建议,最终审批权还是掌握在审评部门。根据试点效果反馈,如反馈情况良好,可以适当扩大试点范围,同时出台鼓励政策推动国内第三方机构的发展,如税收优惠、资金补贴等。医疗器械具有高风险性,第三方机构的准入门槛需设定较高标准,可以从审评人员资质、操作要求、工作的公正性和保密性、防止利益冲突或联结等方面设定第三方机构的认可资质。
3.3.2 出台配套法规,肯定合法地位
利用第三方机构的力量辅助监管部门事务在许多发达国家和地区中屡见不鲜,取得的成效有目共睹。但目前我国医疗器械法规体系尚未明确第三方机构参与监管部门辅助工作的合法地位,这对第三方机构在我国的发展产生了一定的阻碍。建议我国出台对第三方机构进行规范管理的配套法律法规,完善现有法律体系,一方面确定第三方机构合法地位、权利、义务及出现问题时各方应承担的责任;另一方面对第三方的行为进行必要约束和规范,如制订泄露用户信息的惩罚措施等,增强审评的合法性,使医疗器械生产企业可以安心、主动选择第三方机构进行技术审评,提高医疗器械上市速度,推进医疗器械行业快速发展。
3.3.3 加强事前交流,完善沟通机制
FDA十分重视沟通反馈,鼓励制造商与CDRH在医疗器械审评的各个环节开展沟通交流,并将申请人对FDA提出的各种沟通反馈请求统称为Q-Submissions。Q-Submissions包括正式提交申报资料前的资料预提交(pre-sub)、信息报告会、研究风险确定会议、早期正式合作会议、审评过程中的资料提交问题分析会和上市前审批(PMA)100天会议[13]。第三方机构与FDA 之间也采取了邮件、电话等方式进行早期互动。良好的沟通可以大幅提高工作效率,目前的立卷审查制度、特殊产品沟通机制是我国进行审评前移非常有益的探索,建议我国进一步加快和完善沟通交流机制,如建立第三方与省级审评机构、国家安全技术部门专门的沟通渠道,并增设咨询平台,创新沟通方式,除电话和邮件外,可采取定期开座谈会、视频会,创建交流群等方式收集反馈意见,加强注册全过程的交流、答疑及探讨,提高工作效率和质量。
3.3.4 规范审评流程,统一审评尺度
审评尺度不一致是我国医疗器械审评过程存在的主要问题之一[14]。医疗器械涉及科目广泛,不同专业的审评人员对审核产品的理解不同,这可能会造成对同一产品的审核侧重点和补正意见存在不同,使得企业对材料反复修改,延长审评时间。为了确保第三方机构审评工作的顺利实施及审评结果的可靠性,建议相关部门对第三方机构审评流程和审评标准进行统一规定,可以参考其他国家和地区规定的第三方审评流程,编写审评模板,并对审评人员进行统一集中培训,提高审评的正确性,减少重新审查次数。
3.3.5 严格监督机制,完善监管体系
第三方机构作为独立的法人组织,可能存在一定的逐利性。为了保证审评过程的公正性,第三方机构除了保证自身自律,还必须接受监管部门、企业、同行及社会的监督。建议监管部门对第三方机构建立绩效考评机制和动态退出机制,可以根据第三方机构绩效报告、投诉举报及不良事故发生率等情况对其进行考察和评级,并将考评结果及时向大众公布。对于不合格的第三方机构,监管部门可根据结果的严重程度采取问责、处罚直至撤销其审评资格。其次,建议监管部门建立投诉举报机制,将社会对第三方机构审评结果和运营机制的反馈意见作为第三方考评的部分依据。最后,同行成员和新闻媒体等可加大对第三方审评机构的关注,及时曝光不良现象,以起到警示作用。这些措施不仅可以增加公众对医疗器械行业的了解,同时可以督促第三方机构严格遵守工作纪律,保证审评的公正性。