摘要:该文通过查阅官方文件和研究文献,梳理、分析了 FDA 复杂仿制药注册审评过程中的沟通机制,从会议目标、细分类型、申请审核时限和会议程序几方面进行阐述,结合本国复杂仿制药审评审批沟通机制的现状,对两国复杂仿制药审评审批沟通机制进行对比和思考。FDA 复杂仿制药沟通机制分为仿制药上市申请提交前(Pre-abbreviated New DrugApplication,Pre-ANDA)程序、书面咨询和同步科学建议(Parallel Scientific Advice,PSA)程序,通过细分的会议类型充分解决了复杂仿制药研发过程中审评审批层面的难题。建议本国可参考 FDA 做法,探索多样化的复杂仿制药沟通机制,从而提高审评效率,加快具有临床价值的复杂仿制药上市。
在开发新靶点越来越难、仿制药竞争越来越大的情况下,对于无法承担创新转型的仿制药企业,复杂仿制药正在成为一个较为优势的选择[1]。某些复杂仿制药,例如一些吸入或注射产品,即使在药品专利和独占权不再阻碍仿制药获得上市批准之后,其仿制对仿制药企业仍具有一定的挑战性,导致部分仿制药领域缺乏竞争。鼓励复杂仿制药的研发可帮助仿制药企业开拓竞争尚不充分的领域以提高研发回报率。因此,美国食品药品监督管理局(FDA)已建立多种复杂仿制药沟通模式,以解决不同程度的研发技术问题[2—4]。
建立和完善复杂仿制药沟通机制有利于提高复杂仿制药的审评效率,是鼓励复杂仿制药研发的重要途径。然而,本国复杂仿制药的沟通机制建立尚处于初步探索阶段,因此,本文对 FDA 复杂仿制药的沟通机制进行梳理和分析,以为本国复杂仿制药沟通机制的建立提供经验借鉴。
一、美国复杂仿制药审评审批沟通机制的形式
目前 FDA 对复杂仿制药的主要沟通方式有书面咨询、仿制药上市申请提交前(Pre-abbreviated New Drug Application Program,Pre-ANDA Program)程序和同步科学建议(Parallel Scientific Advice,PSA)[5],其中书面咨询主要适用于已发布特定产品指南(Product-Specific Guidance,PSG)的复杂仿制药,且只适用于解决单个问题或一系列密切相关的问题。Pre-ANDA 程序主要适用于未发布 PSG指南的复杂仿制药,解决单独依靠书面咨询不能解决的问题。PSA 程序是为申请人提供同时向欧洲药品管理局(EMA)和 FDA 举办沟通会议的渠道,可同时了解 2 个监管机构对相关技术问题的建议[6—8]。
1.1 Pre-ANDA 程序
2017 年《仿制药使用者付费法案Ⅱ》(Generic Drug User Fee Amendments Ⅱ,GDUFA Ⅱ)中提出了 Pre-ANDA 程序。
1.1.1 Pre-ANDA 程序目标
该程序旨在为申请者向 FDA 提交仿制药上市申请前提供帮助,协助申请者提交完整申请、提高监管机构审评效率、减少复杂仿制药 ANDA 审评周期,以达到提高复杂仿制药可及性的目标。对于 ANDA 申请人而言,Pre-ANDA 程序是一条具有较高价值的信息资源渠道,通过书面沟通和会议可为申请者提供药物研发协助,以及举办提交申请前会议和中期审查周期会议,以帮助阐明产品研发早期和申请审查期间的监管意图。
1.1.2 Pre-ANDA 会议类型
Pre-ANDA 会议类型包括药物研发会议(Product development meeting,RPM)、提交申请前会议(Pre-submission meeting,PSM)、审评中期会议(Midreview cycle meeting,MRCM)。其中,进行 PSM 的要求是应当明确表明潜在ANDA 申请人是否与 FDA 举行了药物研发会议,如果没有召开药物研发会议,潜在 ANDA 申请人应解释为何应当批准召开 PSM。
1.1.3 Pre-ANDA 会议申请审核时限
GDUFA Ⅱ对 3 种类型会议的批准或拒绝设定了期限,具体见表 1。
1.1.4 Pre-ANDA 会议程序
(1)会议申请:复杂仿制药的 RPM 和 PSM 的会议包(Meeting Package)应与会议请求书同时提交给仿制药办公室(Office of Generic Drugs,OGD),而且会议包还应与会议请求一起以电子方式发送到药品评价和研究中心(Center forDrug Evaluation and Research,CDER)。另外会议包应包含与所申请的产品、研发阶段和会议类型相关的信息,以及帮助 FDA 回复申请人提出的问题所需的任何补充信息。
(2)会议请求的评估与答复:OGD 的研究与标准办公室(Office of Researchand Standards,ORS)与药品质量办公室(Office of Pharmaceutical Quality,OPQ)共同确定是否批准 RPM 或 PSM 的请求。FDA 员工负责对会议包进行评估,如对会议包存在疑问可向申请人发送询问信息,申请人可通过信息门户进行回复,评估完毕后会根据 GDUFA Ⅱ批准或拒绝会议请求并向潜在 ANDA 申请人答复。
(3)发布初步书面评论:在复杂仿制药的 RPM 或 PSM 之前,FDA 会召开内部会议讨论会议方案,就对潜在 ANDA 申请人问题的初步答复达成内部一致。对于 RPM,如果 FDA 没有向潜在 ANDA 申请人提供书面答复,FDA 应当在会议前 5 天向潜在 ANDA 申请人的联系人提供初步书面意见。对于 PSM,如果FDA 认为在会议前提供初步书面意见是合适的,则任何此类意见会在会议前 5天通过电子邮件发送给潜在 ANDA申请人的联系人。潜在 ANDA申请人和 FDA之间的会议前沟通包括初步书面评论,可作为讨论的基础,也可作为最终会议的回应。然而,除非潜在 ANDA 申请人和 FDA之间达成协议,否则初步的书面评论不应被解释为最终回应。在收到初步书面评论之后,潜在 ANDA 申请人应提供更新后的议程,并在预定会议前的 48 小时内按优先级顺序提供其讨论的问题列表。需要注意的是,FDA 传达的初步书面评论不应引起新问题,并且会议上也不应遇到新问题。
(4)会议的主持:复杂仿制药的 RPM 和 PSM 会由 FDA 工作人员(通常是ORS主任或指定人员)主持,并以介绍议程声明开始。一般而言,与会者会讨论潜在 ANDA 申请人提出的问题和提供的数据,以协助其制定复杂仿制药的研发计划。分配给 ANDA 的监管项目经理(Regulatory Project Manager,RPM)会主持MRCM,该会议会讨论审查人员或审查团队在审查结束时发现的部分缺陷,以及会议前已向潜在 ANDA 申请人传达的缺陷。
(5)会议过程中申请人的陈述:RPM 过程中通常不需要潜在 ANDA 申请人或 ANDA 申请人进行陈述,因为审查和讨论所需的信息已作为会议包的一部分进行了讨论。如果潜在 ANDA 申请人或 ANDA 申请人计划做演示,申请人应提前与 FDA 讨论,以确保在会议前准备好演示材料,且所有的陈述都应尽量保持简短,从而最大限度地节省时间。会议时长通常不会因发言而增加,但是如果演示文稿中包含了少量与以前数据不同的内容,或新增了未包含在提交给 FDA审查的原始会议包中的数据,FDA 工作人员可能无法对新信息进行及时审查,这可能延长会议时间。另外,FDA并不要求参加 MRCM的申请人进行任何演示[9]。
(6)会议纪要:在会议结束前,FDA 参会者和潜在 ANDA 申请人或 ANDA申请人应总结讨论要点和相关协议要求。一般情况下,FDA 会要求潜在 ANDA申请人或 ANDA 申请人提交会议纪要以确保对会议结果和行动项目达成相互理解。FDA 工作人员可在会议记录中添加或进一步澄清总结中未涉及的要点,总结可以在会议结束时完成,也可以在讨论每个问题后完成。
1.2 书面咨询
FDA 在 GDUFA Ⅱ承诺函[10]中提出,对于复杂仿制药,FDA 会对标准的书面咨询和复杂的书面咨询进行审查,并做出有意义的回答,为药物研发或监管决策提供信息。书面咨询适用于解决单个问题或一系列密切相关的问题,在《有关仿制药研发的书面咨询》(Controlled Correspondence Related to GenericDrug Development)[11]指南中提出,FDA 会在提交日期后 60 天内审查并回复 90%的标准书面咨询,在提交日期的 120 天内审查并回复 90%的复杂书面咨询,并在收到请求后的 14 天内澄清书面咨询回复中的歧义。
1.3 PSA 程序
FDA 与 EMA 合作启动了一项试点计划,为 FDA 复杂仿制药产品的 ANDA和 EMA 混合产品的上市授权申请(Marketing Authorization Applications,MAAs)提供科学建议[12]。
1.3.1 PSA 程序目标
PSA 程序的目标是为 EMA 和 FDA 提供一种机制,与申请者共同交流双方机构对混合/复杂仿制药产品研发阶段科学问题的看法。PSA 程序预计从混合/复杂仿制药产品的生命周期开始就增加双方机构与申请者之间的沟通,成功的合作可能会使申请人更深入地了解双方的监管决策基础,优化申请人的产品研发计划,并帮助申请人避免为了满足双方机构需求而进行不必要的研究。
PSA 程序的重点是分享信息和观点,潜在优势是实现双方机构的协调和融合,申请人可使用 PSA 程序来确定研究设计是否可能为 2 个监管机构接受,可能受益于 PSA 程序的研究包括涉及创新生物等效性研究设计以及使用建模和模拟等方法的比较性非临床和临床研究。在进行 PSA 会议之后,申请人会对双方机构各自的监管要求和所讨论的研发项目科学建议有更清晰的了解,如果 2 个机构的建议有分歧,申请人会清楚地了解产生分歧的原因。FDA 还公布了 EMA和 FDA 达成一致的试点项目一般原则,将使 PSA 程序的过程和目标更透明,并有助于回答有关该程序的问题。PSA 会议的申请将根据工作量、工作人员的可获得性和 PSA 程序的预期价值而获得批准。对于每个申请,各机构会关注具体发展问题与申请者举行一次三方会议,三方会议一般为 1.5 h,但时间会根据会议要讨论的问题数量和复杂性而延长。
1.3.2 PSA 程序会议流程
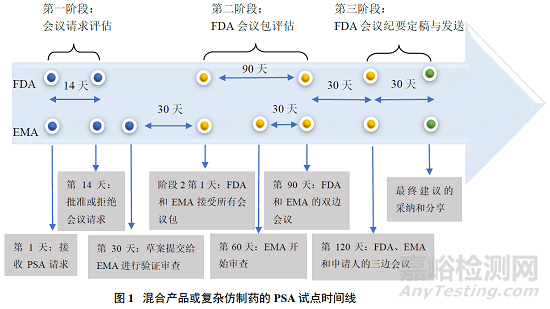
2021 年 9 月 15 日,PSA 程序试点开始。试点 PSA 会议流程主要可分为 3 个阶段,申请人请求与 EMA 和 FDA 展开会议、PSA 会议准备与召开和 EMA 和FDA 向申请人传达书面答复,下面分阶段详细论述。
(1)第一阶段——申请人请求与 EMA 和 FDA 展开会议
一般而言,提交 PSA 会议要求的申请人应将重点放在涉及混合产品或复杂仿制药研发的特定科学问题上,希望从 EMA 和 FDA 获得进一步的科学建议。另 外 PSA 申 请 人 应 当 通 过 emainternational@ema.europa.eu 向 EMA 和atpreANDAHelp@fda.hhs.gov 向 FDA 发送请求进行 PSA 的信函(论证函),以启动 PSA 会议。
提交 PSA 申请并不能保证 PSA 会议得到批准,由于各种原因双方机构可能会拒绝参加这样的会议。如果申请人的 PSA 会议申请未获批准,申请人可按照各机构的正常程序,单独向各机构咨询,在协商咨询过程中,PSA 申请人会邀请任何一方一定数量的专家参加另一方机构的讨论。但是,申请人不应同时通过 PSA 和 FDA 的 Pre-ANDA 程序提交相同的会议请求。
(2)第二阶段——PSA 会议准备与召开
如果 2 个机构都批准了申请人的 PSA 请求,则申请人将按照各机构的电子邮件内容确认该协议并指定各机构的主要联系人。申请人应在提交完整会议包30 天前通过 IRIS 门户(https://iris.ema.europa.eu/)向 EMA 提交会议包草案以进行验证审查和修订。
一旦会议包草案由 EMA 验证通过,申请人应通过 IRIS 门户向 EMA 和通过preandahelp@fda.hhs.gov 向 FDA 提交最终的完整会议包。在完整的会议计划中,申请人应提供研发计划的简要历史和产品研发状态讨论的问题列表,根据主题分组并对每个问题明确编号。申请人应对每个问题的背景、目的,以及任何支持的理由或数据进行简要说明,而且会议包应放在所有问题的摘要列表之后,每个问题后都应有相应的理由、基本原理或数据支持。
PSA流程的第2阶段通常对应于FDA的Pre-ANDA前会议的120天时间线,覆盖了 EMA 科学建议工作组(Scientific Advice Working Party,SAWP)为其科学建议流程规定的 70 天时间线,因此这 2 个机构目前确定的举行科学会议的时间表可进行适当调整,以与双方机构制定的时间线保持一致。2个机构的主要联系人会商定 EMA 和 FDA 的双边会议和与申请人的三边会议的时间,并达成共识,然后与申请人共享三边会议的时间。双方机构会在三边会议举行前进行双边电话会议或视频会议,以进一步讨论申请人提出的问题。之后,申请人会与 EMA和 FDA 一起参加三边 PSA 电话会议,双方机构可在三边会议之后与申请人进行电话或视频会议。
申请人和双方机构的电话会议或视频会议通常安排在申请人提交完整会议包后 120 天左右。EMA 问题清单和 FDA 的初步答复会在三方会议前约 14 天公布,各机构的指定主要联系人会与申请人协调会议的后续工作。
(3)第三阶段——EMA 和 FDA 向申请人传达书面答复
在经过 PSA 会议之后,各机构会保留其关于药物研发和上市申请的单独监管决策。在联合讨论后各机构的建议可能仍然不同,因此各机构会根据通常的程序和时间就 PSA 会议期间提出的问题向申请人提供独立的建议。参与 PSA 程序不应被理解为确保任何一方机构对上市申请采取任何行动,双方机构将努力提供相似的 PSA 回应,但是双方机构仍会致力于满足各自的会议进程和时间。另外 PSA 程序不应对任何一个机构满足其个人业绩期望的能力产生不利影响,因此,这 2 个机构都会承诺已清楚认识到对方的国内业绩预期,并在安排 PSA会议时尽可能表现出灵活性。
根据上述 PSA 程序 3 个阶段的概述,可总结出 EMA 与 FDA 的混合产品或复方仿制药的 PSA 试点时间表,如图 1 所示。
二、本国复杂仿制药审评审批沟通机制演变与建立
2016 年 6 月 2 日,原国家食品药品监管总局发布的《药物研发与技术审评沟通交流管理办法(试行)》[13]中提出规定的沟通交流会议优先适用于创新药物、采用先进制剂技术药物,以及临床急需药物研发的注册申请的沟通交流。未明确提到复杂仿制药。2020 年 12 月 11 日,国家药品监督管理局药品审评中心发布了《药物研发与技术审评沟通交流管理办法》的通告(2020年第 48号)[14],提出Ⅲ类会议包括复杂仿制药、一致性评价或再评价品种就重大研发问题(参比制剂的选择、生物等效性的评价标准等)提出的沟通交流申请。Ⅲ类会议一般安排在申请后的 75 天内召开。虽然本国在药物研发沟通机制的探索中逐渐开始重视复杂仿制药,但是相较 FDA,本国复杂仿制药沟通交流机制的相关规定仍不够全面细致,特别是对相关主体的职责以及一些流程化的规定仍需进一步完善。
三、结语
本国针对复杂仿制药统一以Ⅲ类会议来处理,为统一的时限。FDA 专门针对复杂仿制药细化设置了 3 种不同的会议类型,适用于不同的情境,并根据不同会议类型分别设置时限,且通过细分的会议类型,FDA 复杂仿制药沟通机制可充分解决复杂仿制药研发过程中的技术难题,以鼓励复杂仿制药的研发,对本国的复杂仿制药审评沟通交流机制有诸多启示。目前,本国复杂仿制药特定指南尚不完善,注重复杂仿制药的研发各阶段的沟通交流机制的建设尤为重要,可参考、借鉴 FDA 在沟通机制方面的经验,专门建立复杂仿制药的沟通交流机制,探索多样化的复杂仿制药的沟通机制,以提高审评效率,加快具有临床价值的复杂仿制药上市。