您当前的位置:检测资讯 > 行业研究
嘉峪检测网 2025-04-13 11:31
2025年4月10日的仿制药论坛,FDA的Darby Kozak博士发表了关于仿制药批准的缺陷分析。
基于仿制药企业规模(企业获批ANDA数量)和仿制药产品复杂度的近期简化仿制药(ANDA)提交分析(2018至2023财年)、批准率及主要缺陷趋势:
• 局部皮肤科药物ANDA的主要生物等效性(BE)缺陷及常见BE缺陷的详细分析
• FDA为解决共性问题并促进更多首轮和第二轮ANDA获批推出的行动计划与专项计划。

大约38-40%的2018财年和2022财年仿制药申请(ANDA)是在第二轮审评周期内获得批准的(申请人此前已收到一封完整回应函(CR)后获批)。美国食品药品监督管理局和仿制药行业应努力提高第二轮审评周期内获批的ANDA比例。
2018财年和2022财年,复杂产品分别约占ANDA提交量的14%和17%。这些复杂产品ANDA中约25%在第二个评估周期内获得批准。
2018财年和2022财年提交的ANDA材料已获得至少两个评估周期的充分审评时间。大部分2018财年提交的ANDA应已接近最终处理状态(如获批或撤回/放弃),而2022财年在审评中(CR)的ANDA多数将进入第三轮评估周期。

约300家不同企业在2018财年及2022财年提交了仿制药申请
• 特大型企业(递交100件以上ANDA)、大型企业(递交20至100件以下ANDA)、
型企业(递交6至19件以下ANDA)、小型企业(递交6件以下ANDA)
• 特大型企业递交了半数仿制药申请。
• 复杂产品ANDA占特大型和小型企业递交总量的18%。大型和中型企业递交的ANDA中有10%为复杂产品。
无论企业规模大小,生产与药品成品缺陷始终是第一轮审评函中最常见的主缺陷项。

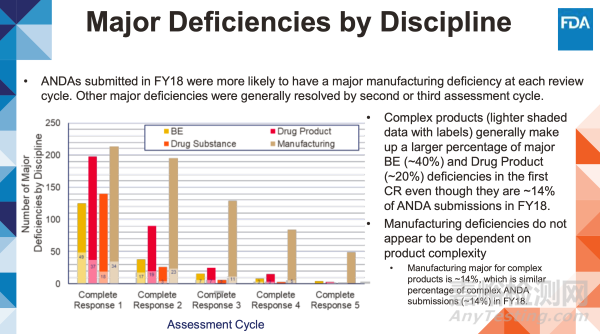
在2018财年提交的仿制药申请中,每个审评周期出现重大生产缺陷的比例更高。其他主要缺陷通常在第二或第三评估周期得到解决。
• 复杂产品(颜色较浅并带有标签的数据)尽管仅占2018财年ANDA申报量的14%,却在首次完整回复函中构成了生物等效性缺陷(约40%)和制剂缺陷(约20%)的主要部分。
• 生产缺陷的出现似乎与产品复杂程度无关
验证数据不足:
生物分析方法
– 样品稳定性、基质变化或方法的相关信息可能不适用(如定量下限LLOQ、色谱图质量差)
体外生物等效性(BE)研究:
体外释放试验(IVRT)/体外渗透试验(IVPT)/体外结合研究
– 支持体外释放/渗透测试的方法开发和验证信息(如区分能力、实验条件的合理性),或实现最大组合


• GDUFA III倡议和在整个仿制药流程中加强沟通的机会,以帮助识别(潜在)重大缺陷的解决方案。
• 沟通交流、开发会议、学科审查函(DRL)、信息请求(IR)、中期审评会议、CR后会议等。


结论:
• 大约40%提交的ANDA在第二个评估周期获得批准。其余60%的ANDA仍存在未解决的缺陷,需要更多评估周期且无法获得批准。
• 生产(主要与设施相关)、药品、生物等效性和原料药是首次评估周期中发现主要缺陷最多的方面
• 最常见的主要生物等效性缺陷(38%)源于体外研究不足
体外释放试验(IVRT)和体外渗透试验(IVPT)研究不足,是采用基于特性生物等效性方法的局部皮肤科药物ANDA最主要生物等效性缺陷
• FDA和GDUFA III计划通过加强申请人沟通,提升了ANDA评估效率并减少了常见缺陷
• 产品特定指南、受控函回复、GDUFA会议、学科审评信、信息请求、研讨会和网络会议
• 自2018财年以来收到超过175个局部皮肤科药物ANDA其中多数已获批上市


来源:文亮频道


