您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-05-11 11:26
摘 要 / Abstract
药品上市许可持有人(MAH)与药品生产者可以是不同主体,实行持证和生产分离管理,是MAH 制度的基本内容。我国既往是实行持证和生产合一的管理制度,要求MAH 必须是该药品的生产企业。随着制药产业的发展和监管科学的进步,持证和生产合一管理制度下生产设施重复建设、药品主体责任落实不充分等问题日益突出,亟需建立和实施MAH 制度,以持续强化MAH 责任,合理调整实施持证和生产分离管理。本文着眼于行业需求反映突出的持证和生产问题,针对MAH 跨境持证和生产管理、境外MAH 的境内代理人及其职责管理、MAH 与原料药持证和生产管理3 个方面,分析当前监管措施和行业需求,剖析监管与需求之间的差异和难点,并针对这些差异和难点,对比分析了欧盟、美国、日本和我国的相关监管措施及监管特点、差异和成效,以期为完善我国MAH 制度提供参考与借鉴。
The marketing authorization holder (MAH) may or may not be the same as the drug manufacturer. The separation between the holder and the manufacturer is one of the essential aspects of the MAH system. Under the previous regulatory system in China, the holder-manufacturer-in-one restriction was implemented, requiring the MAH to hold the certificate of a drug product. With the development of the pharmaceutical industry and regulatory science, issues such as duplicate construction of production facilities and insufficient definition and enforcement of responsibilities of the drug holder under the holder-manufacturer-in-one system have become increasingly critical. It is necessary to establish the MAH system to strengthen the responsibilities of the MAH and the separation between the holder and the manufacturer. This article focuses on the key issues of certificate holding and product manufacturing reflected by industry demand, especially the current regulatory approaches and industry expectations on cross-border certificate holding and product manufacturing under an MAH. It also examines the foreign MAH-delegated local agent and its roles and responsibilities, as well as the relationship among the MAH, drug substance holder, and drug substance manufacturer. The gaps between supervision and demand, as well as challenges, are analyzed. To explore the reference for the implementation of China's MAH system, this article compares key points, differences, and performance of various regulatory measures in Europe, United States, Japan and China.
关 键 词 / Key words
药品上市许可持有人;上市许可;生产许可;持证和生产
marketing authorization holder; marketing authorization; manufacturing authorization; certificate holding and product manufacturing
药品上市许可持有人(marketing authorization holder,MAH)制度是欧洲、美国、日本等国家和地区药品监管领域的通行做法[1]。尽管各国和地区的药品监管制度设计及实施有所不同,但欧盟、美国、日本药品监管均以MAH 承担药品全过程、全生命周期主体责任为基础,持有药品上市许可证明文件的权责与药品生产许可下的药品生产活动(以下简称持证和生产)可以由不同主体承担,即实行持证和生产分离管理。在MAH 制度下,MAH 作为药品质量安全的责任主体,可优化资源利用,将药品生产交由自有生产场地或受托药品生产企业完成,以提高新药研发效率和保障上市药品持续供应。
我国既往要求MAH 必须是该药品的生产企业,即持证和生产合一管理。在当时的行业状况和监管制度下,持证和生产合一是药品质量安全监管的风险底线,即药品生产企业具备一定的质量管理体系、具有履行管理职能的人员组织基础及可追责对象。但随着制药产业的快速发展,持证和生产合一管理下的生产重复建设问题日益凸显。一方面制药产能大量闲置,现有生产资源得不到充分利用;另一方面新药研制企业又需为了自有生产设施设备而重复建设,占用了大量资金和资源。同时,随着药品监管科学的不断进步,以药品生产许可为风险底线的做法,也不足以充分落实药品全过程、全生命周期主体责任。
为适应药品行业的发展和监管制度的完善,我国亟需建立实施MAH 制度,在持续强化MAH责任的同时,合理调整、实施持证和生产的分离管理。2015 年,《全国人民代表大会常务委员会关于授权国务院在部分地方开展药品上市许可持有人制度试点和有关问题的决定》发布,决定在部分省(自治区、直辖市)开展MAH 制度试点工作[2]。经过几年的试点工作及相关经验的积累,2019 年新修订的《药品管理法》发布,我国全面实行MAH 制度[3],并明确了MAH 可以自行生产也可以委托药品生产企业生产药品。持证和生产分离管理,对鼓励医药领域创新,促进产业资源优化配置,落实各方责任方面均起到积极的作用。
在处理持证和生产相互关系方面,我国MAH 制度的相关法律法规明确了在提交境内生产药品的上市许可申请、上市后生产场地变更的补充申请、上市后MAH 变更的补充申请时,申请人和生产企业应当已取得相应的药品生产许可证[4-5] ;明确了境内MAH 和境内药品生产企业均需获得药品生产许可证[6]。这些监管措施的确定和调整充分考虑了与既往制度的衔接,理顺了相关行政流程和行政许可要求。但仍存在涉及持证和生产的关键问题有待解决,包括跨境问题、多生产场地分段生产问题等,在此基础上又产生了新的关键问题,例如境外MAH 的境内代理问题、MAH 药品生产许可证管理问题、MAH 与原料药持证和生产管理问题等。在探索和研究上述问题的解决办法时,其他国家和地区的成熟经验具有重要参考价值。本文就欧盟、美国、日本的相关经验对我国MAH 制度实施中涉及持证和生产的一些关键问题的启示与借鉴进行了探讨。
1、MAH 与药品生产场地分属境内外的管理
我国全面实施MAH 后,MAH 可以委托其他生产企业生产药品,以实现持证和生产分离管理。但目前该分离管理仅适用于MAH 与药品生产场地同属境外或者同属境内的情形。当持证和生产分属境内外,即“境内持证、境外生产”或“境外持证、境内生产”时,现有制度中尚无可适用该情形的申报路径。
我国对境内生产药品和境外生产药品的监管存在较多差异,本文进行了列举,见表1。MAH和生产场地分属境内外时,会产生持证和生产的跨境交叉,而现有的监管措施尚较难交叉适用。在现有的监管框架下,除了境外生产的药品待包装制剂半成品在境内生产场地分包装按备案管理这一特殊情况外[7],我国尚不接受其他情形的MAH 跨境持证和生产。
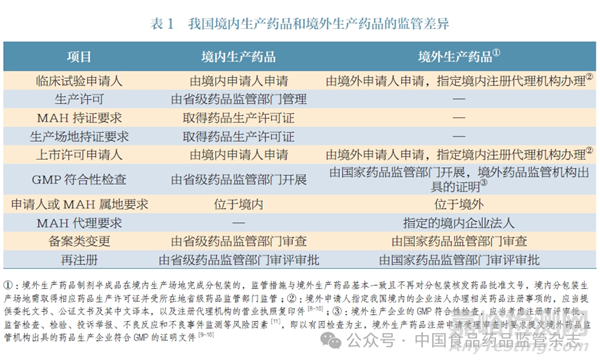
在欧盟、美国、日本的监管制度下,MAH 持证要求不因药品生产场地在境内或境外而有所区别。本文对欧盟、美国、日本和我国的MAH 持证要求进行了比较,见表2。尽管在MAH 持证要求方面有差异,但欧盟、美国和日本接受跨境持证和生产,尤其是“境内持证、境外生产”情形。
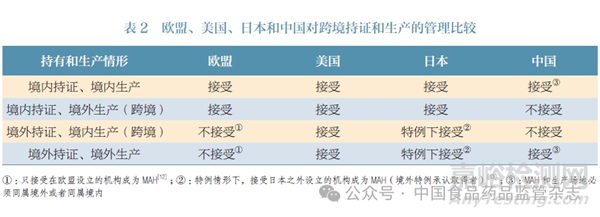
总体而言,欧盟、美国、日本MAH 制度能够接受跨境持证和生产,而不将其视为不受控的监管风险,即基于其监管体系能够良好地落实MAH 主体责任,境外生产和境内生产也都能够受到严格的监管,本文就其具体做法进行了介绍。
1. 欧盟
MAH 主体责任落实方面,在欧盟的成员国( 以下简称成员国)境内或境外生产的药品,其MAH 都是成员国境内的机构,且无论药品在成员国境外或者境内生产,均要求MAH 授权符合欧盟资质要求的质量受权人(qualified person,QP) 负责药品的上市放行[12]。生产监管方面,成员国的药品监管部门应确保本国辖区内药品生产已获得相应生产许可(manufacturing authorisation), 成员国之外生产的药品则由MAH 向其所在成员国申请生产进口许可(manufacturing import authorisation,MIA),并受成员国监管部门的GMP 监管。欧盟的监管措施与其监管框架相适应,充分利用欧盟委员会(European Commission,EC)、欧洲药品管理局(European Medicines Agency, EMA)和各成员国的监管体系和资源。例如,EMA 和各成员国监管部门对药品上市及上市后监管中,分别履职并采取相应的监管措施,以此控制药品的全过程、全生命周期的监管风险,见表3。
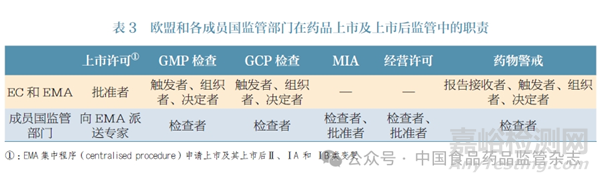
欧盟MAH 制度对我国具有一定的借鉴意义。例如, 规定MAH 必须是欧盟地区内注册的机构,更有利于属地MAH 承担相关的主体责任,有利于责任界定,也有利于监管部门对MAH 进行监督的相互沟通。而欧盟监管制度以各成员国的监管体系及其相互协调为基础,形成集中与分散并存的监管机制。相较而言,我国省级药品监管部门在事权、资源、相互协调等方面尚显不足,尤其在MAH 持证监管和生产监管方面,国家与省级之间、省级与省级之间的沟通和协调方面。可考虑部分借鉴欧盟的管理方式,加强省级药品监管部门的事权和职能,对跨境持证和生产的情形,建立国家药品监管部门集中管理、省级药品监管部门分散管理以实现相互支撑和协调。
2. 美国
在MAH 主体责任落实方面,美国的对应主体一般称为申请持有人(application holder)或证书持有人(license holder),美国《联邦食品药品和化妆品法案》(Federal Food, Drug and Cosmetic Act)第355 条规定, 任何人均可提交有关任何药品的上市申请。因此,美国境内或境外的机构均可成为MAH。在生产监管方面,无论是境外还是境内的生产场地,美国均实行生产设施登记制度(establishment registration)管理[14-15],不颁发药品生产许可证或GMP 证书。美国食品药品监督管理局(Food and Drug Administration,FDA)在新药上市许可审评期间,基于审评需求开展批准前检查(pre-approval inspection,PAI 或者pre-license inspection,PLI), 这些问题必须在批准申请之前解决。若未能通过PAI/PLI,或在检查中发现不符合现行药品生产质量管理规范,将不予批准药品上市许可申请。无论生产场地在美国境内还是境外,FDA 均会对生产场地进行检查。对于境外的生产设施,可由FDA 在境外国家和地区设立的办公室工作人员(临时工作人员或专职工作人员)进行检查。此外,FDA 与欧盟、英国、瑞士等国家和地区达成互认协议(mutual recognition agreement,MRA),认可境外监管部门就认可范围内的药品开展的符合美国要求的检查。目前,MRA 认可范围不包括人用疫苗、人血液来源药品、人血浆来源药品、人组织器官来源药品、先进治疗药品(advanced therapy medicinal products)、临床试验用药等。此类药品有待后续评估以决定是否扩展互认协议的认可范围[16]。
与欧盟集中- 分散并存互补的机制不同,美国的MAH 制度是由FDA 统一集中管理,其良好实施的基础是FDA 强大的全球监管延伸能力,无论境内还是境外的MAH 和生产场地均受FDA的监管检查,以确保监管的风险可控。FDA 在境外设立检查办公室开展境外检查工作以及与其他国家和地区达成检查互认等举措,对我国加强MAH 持证和生产的监管也具有一定的借鉴意义。
3. 日本
日本监管部门从企业责任体系、药品有效性安全性、药品生产方法和管理体系3 个方面,建立了多维度的药品审查体系[17]。企业责任体系方面, 申请人拟成为MAH 时,需提出生产销售从业许可申请并获得相应许可,以确保MAH 有能力管理药品的质量和上市后安全。药品有效性安全性方面,除了对临床试验申请、上市许可申请的审评批准外, 药品从研发到上市的过程中以可靠性保证即药物非临床研究质量管理规范(good laboratory practice,GLP) 调查、药物临床试验质量管理规范(good clinical practice,GCP)调查、上市后研究质量管理规范(good post-marketing study practice,GPSP) 调查确认其申请资料的可靠性, 以符合性调查即药品生产质量管理规范(good manufacturing practice,GMP)、再生医疗等制品制造管理和质量管理规范 (good gene, cellular,and tissue-based products manufacturing practice,GCTP) 确定药品生产合规[18]。药品生产方法和管理体系方面,申请人自有生产场地或者委托生产企业均需提出生产从业许可申请并获得许可,境外生产场地还应递交境外生产企业认定申请;在药品上市申请审评过程中还需提出GMP 符合性检查申请并通过书面或现场检查。
日本MAH 制度的建立和实施情况对我国有更多借鉴之处。以2005 年修订实施的《药事法》为指引,日本将监管制度调整为生产许可与上市销售许可分离管理,持证的MAH 不必是该药品的生产企业,该历程与我国正在经历的MAH 制度建立情形相似。日本多维度的审查体系和流程、药品开发上市全程可靠性保证与符合性调查等措施,对我国基于已有监管基础建立完善MAH 制度具有较多可借鉴之处。

2、境外MAH 的境内代理人及其职责管理
如前所述,我国尚不接受跨境持证和生产,即境外生产的药品必须由境外MAH 持有,境内生产的药品必须由境内MAH 持有。境外MAH 应当指定境内企业法人,作为境内履行MAH 义务、承担MAH 连带责任的主体[3]。但该境内企业法人(即境外MAH的境内代理人)与持证和生产的关系尚不够清晰,境外MAH 和境内代理人在境内和境外的主体责任和连带责任尚不够明确,这也是MAH 不能实施跨境持证和生产的另一限制因素。
在境外MAH 代理人承责履职方面,现有法律法规中明确规定由境外MAH 指定的境内代理人负责的事务包括:①履行医药代表备案和管理责任[19] ;②履行年度报告义务[20] ;③代为填报短缺药品生产供应及停产报告信息,并在线提交至境内代理人所在地省级药品监管部门[21] ;④按规定组织实施在境内的药品召回[22] ;⑤具体承担进口药品不良反应监测、评价、风险控制等工作[23] ;⑥向进口口岸所在地省级药品监管部门报告进口疫苗在流通管理过程中发现的可能影响疫苗产品质量的重大偏差或重大质量问题[24] ;⑦办理批签发[25] ;⑧代为撰写疫苗质量年度报告[26] ;⑨协助药品监管部门开展对产品境外研制、生产场地的检查和违法违规行为的查处等[11]。但对于境外MAH指定的境内代理人准入资质、指定程序、承责履职及其监督管理等,我国尚未制定发布专门的监管规章予以系统完整的规定。
与我国一样, 欧盟、美国、日本等国家和地区对境内MAH均不设代理人,责任界定明确,即境内MAH 是药品全过程、全生命周期的单一责任主体。但在是否准许境外机构作为MAH 以及其境内代理人的管理方面,欧盟、美国、日本等国家和地区各有不同规定,见表4。相较而言,我国与日本的监管要求更为相似。

1. 欧盟
欧盟只接受在其成员国境内设立的机构成为MAH,不存在成员国境外MAH 的境内代理人情形。值得关注的是,欧盟的MAH为单一主体,其规定若有欧盟成员国内的多个申请人属于相同母公司或集团,且这些申请人就相关药品的市场投放已达成协议或开展协同行动的, 须视为同一MAH[27]。该项规定对我国处理集团公司下多个主体申请药品上市情形的责任界定和监督管理具有借鉴意义。
2. 美国
美国境外或境内的任何人均可提交有关任何药品的上市申请并成为该药品的MAH。境外主体担任MAH 时, 境外MAH 必须指定单一的境内代理人(United States agent), 作为主要和(或)默认联系人负责与FDA 联络。值得关注的是, 该境内代理人不承担境外MAH 的连带责任。MAH 对药品负有第一责任,FDA 会审查MAH 的境内代理人的相关责任,具体责任界定取决于境内代理人与MAH 之间达成的协议规定的职责划分。美国《联邦法规汇编》(Code of Federal Regulation) 第21 条207.69 规定,境内代理人必须在美国境内居住或维持营业场所,不得是邮箱、答录机或服务机构,也不得是该代理人实际不在场的其他场所。代理人负责:①审查、传播、传递信息和回应FDA 与境外申请人的所有沟通,包括紧急沟通;②回答有关进口或提供进口到美国药品的问题;③协助FDA 安排检查;④如果FDA 无法直接或迅速联系境外申请人,FDA 可能会要求其在美国的代理人提供信息和(或)文件,FDA 向美国代理人提供信息和(或)文件等同于向境外申请人提供相同的信息和(或)文件[28]。无论是境内MAH还是境外MAH,都是其在美国上市药品的单一责任主体。
在美国的制度下,一家境外MAH 与FDA 联络,指定单一境内代理人是恰当合理的。但在我国MAH 制度要求下,境内代理人负责履行MAH 上市药品的各项义务,若是一家境外MAH 持有多款药品且各药品的市场准入环境、商业和经营合作方式等差异巨大,则按有关法律法规及相关草案[29] 拟指定单一境内代理人, 履行规定的责任可能存在困难。
3. 日本
无论生产场地在日本境外还是境内,通常情况下,MAH 都应为在日本境内设立的机构。特例情形下,非日本设立的机构可成为境外特例MAH,则需指定日本境内拥有同类药品生产销售许可证的指定代理MAH 实现药品在日本的上市,并接受监管部门的监管,承担药品上市责任[13]。代理MAH 必须具备质量管理和药物警戒管理的人员、组织和履职履责能力,且不能委托给他人。日本的MAH 制度对境外MAH指定的境内代理MAH 按照许可证管理,由境内代理MAH 实际履行MAH 责任,最大程度确保药品开发和上市相关责任由具有相应资质并受日本监管部门完整监管的境内企业承担,对我国建立完善境外MAH 的境内代理人管理体系更具参考意义。
3、MAH 与原料药的持证和生产管理
我国MAH 制度下,MAH 与原料药持证和生产关系的问题也有待解决。在MAH 试点时,曾将原料药纳入MAH 试点范围[30],即原料药注册证书持有者可以委托其他生产企业生产。但随着原料药管理由注册制度调整为登记及关联审评审批制度,不再发放原料药批准文号[31],不再单独受理原料药注册申请[32],原料药由持有相应药品生产许可证的生产企业登记且不得委托生产[6],意味着在原料药的管理方面,持证(化学原料药批准通知书)[4] 与生产不能分离。然而随着专业化、分工化、跨地域资源协作整合的生产模式日趋发展成熟,与其他制造领域一样,医药领域对生产资源的灵活利用有强烈需求。相关企业也希望通过委托生产等方式,将产品生产活动分配于多个生产场地,以最大程度实现生产运营。
现行监管措施下,若不能自产自供原料药,则MAH 必须对外部供应的原料药质量承担责任[31],但又不能持有该原料药的行政许可,对于创新药品的原料药研制生产而言,管理困难和产权担忧更为突出。虽然MAH 拥有原料药的知识产权,但自产自供原料药的资源投入大、研制进展慢、利用效率低,通常需要委托外部生产企业生产原料药。限于原料药必须由原料药生产企业登记和持有,MAH 只能通过协议与原料药生产企业约定供应等事项,存在较大的商业风险;并且不同于仿制药品,创新药品没有更多可供选择的原料药商业供应来源,持续供应的风险更高;当MAH拟变更原料药生产企业时,不但需要重新登记原料药并等待原料药与制剂的关联审评审批,也缺乏行政途径收回由原先的生产企业持有的原料药行政许可。因此,该领域迫切希望灵活利用外部生产资源生产原料药的同时,MAH也能够持有原料药行政许可,以充分保护自身权益,降低风险。
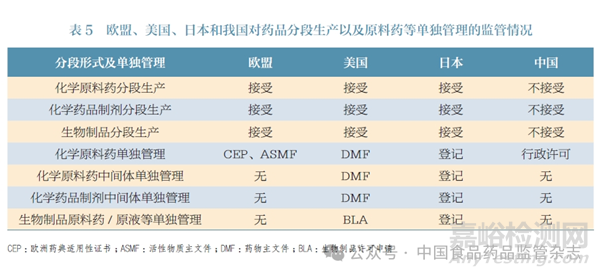
现行制度下的另一较为突出问题,体现在原料药登记持证范围仅限于化学原料药,而化学原料药中间体、化学药品制剂中间体、生物制品原料药/ 原液没有单独登记持证的实施途径。具体限制性要求体现为:①化学原料药全部生产过程在同一生产场地完成;②化学药品制剂全部生产过程在同一生产场地完成;③生物制品从原液至制剂的全部生产过程在同一生产场地完成。不能以委托生产方式将化学原料药、化学药品制剂、生物制品的各段生产分配于不同生产场地。
欧盟、美国、日本等国家和地区的药品监管机构未对化学原料药采取单独的行政许可,但有非强制的单独管理方式。化学原料药所有权人可以选择相应的单独管理,也可以选择由MAH 将化学原料药资料纳入药品注册资料中提交。本文总结了欧盟、美国、日本和我国对药品分段生产以及原料药等单独管理的监管情况,见表5。可以看出,欧盟、美国、日本的药品注册接受MAH 以委托生产方式将化学原料药生产交由其他生产企业,并在药品注册资料中列明。此外,欧盟、美国、日本接受化学原料药、化学药品制剂、生物制品的各段生产分配于不同生产场地,并且美国和日本对化学原料药中间体、化学药品制剂中间体、生物制品原料药/ 原液等也有单独管理的方式。
1. 欧盟
在欧盟,化学原料药的资料可以纳入制剂的注册资料中一并提交,也可以按照欧洲药典适用性证书(certificate of suitability to monograph of European pharmacopoeia,CEP) 认证或按照活性物质主文件(active substance master file,ASMF)注册以形成原料药资料的单独管理;若化学原料药涉及多个生产场地的分段生产,需在原料药的资料中列明各中间体的所有生产场地。CEP 证书和ASMF 注册均不适用于生物制品原液,欧盟的监管体系也不对化学原料药中间体、化学药品制剂中间体、生物制品原料药/ 原液进行单独管理,由MAH 对药品生产的全过程包括分段生产及其产物负责。
2. 美国
美国建立了药物主文件(drug master file,DMF)制度,该制度的实施对象广泛,化学原料药、化学原料药中间体、化学药品制剂中间体等均纳入DMF归档的范围。值得注意的是,生物制品原料药/ 原液等分段生产产物不在DMF 归档范围内。在特定情况下,不同MAH 的生产企业以合作生产的方式分段生产生物制品,则采取注册批准制度进行管理。例如,甲公司生产的生物制品原料药/ 原液、中间体用于乙公司的后续生产的,甲公司提出生物制品许可申请(biologic license application,BLA),获得批准且在“适应症(indication)”项下注明“ 用于乙公司后续生产(for further manufacturing use)”,使之能用于乙公司的后续生产,并以此形成对生物制品分段生产产物的单独管理[33]。
3. 日本
2005 年,日本《药事法》修订后,在引入和实施MAH 制度的同时,建立了日本的原料药等登记制度[34],该制度近似于欧盟ASMF 和美国DMF 的融合,整体上更趋近于DMF 管理,但其可登记范围则相较美国DMF 更为广泛。不仅可登记化学原料药中间体、化学药品制剂中间体、生物制品原料药/ 原液(例如肝素钠、胰岛素、胰高血糖素、透明质酸酶、依库珠单抗等)[35],还可登记生产再生医疗制品所用原材料(例如细胞、培养基、培养基添加物、细胞加工用物料等)。日本的原料药等登记制度与美国DMF 制度都更多地从有利于保护物料供应商的商业秘密角度,允许单独/ 分段生产这些物料的供应商将技术资料直接提供给监管部门,用于支持使用该物料的药品注册申请。
由上可见,尽管欧盟、美国、日本等国家和地区的原料药管理方式有所不同,但均支持化学原料药的委托生产,也支持化学原料药、化学药品制剂、生物制品的各段生产分配于不同生产场地。对分段生产的产物(化学原料药中间体、化学药品制剂中间体、生物制品原料药/ 原液等)是否单独管理,欧盟、美国、日本等国家和地区有各自考虑和做法。鉴于我国已有对化学原料药采取单独管理(登记及关联审评审批的行政许可)的经验和基础,笔者认为美国和日本对分段生产产物单独管理的措施更具借鉴意义,若能将单独管理的范围扩展到药品分段生产的产物,对于降低监管风险以及接受各种方式的药品分段生产具有积极意义。
4、结 语
持证和生产问题,是制定和实施我国MAH 制度的初始动力之一,也是调整和完善MAH 制度的难点之一。推进持证(药品上市许可)和生产(药品生产许可)分离管理,需要在既有监管基础上,进一步解决境外生产药品和境内生产药品的监管差异,以适应制药行业全球化和监管科学国际协调的发展需求;落实MAH的责任履职要求并采取对应的监管措施,以保障持证和生产分离管理下的药品质量安全;结合我国监管特点例如化学原料药按药品管理等情形,充分实现持证和生产的分离管理。
通过对欧盟、美国、日本等国家和地区监管情况进行分析,对比各国和地区实现持证和生产分离管理的制度基础、监管历程、具体措施及其成效,能为我国MAH 制度的持续完善和难点突破,提供具有借鉴意义的工作经验和研判依据。例如,在药品MAH 持有药品生产许可证方面,日本对MAH 的企业责任体系的生产销售从业认证可能更具参考价值;在MAH 跨境持证和生产方面,欧盟、美国、日本等国家和地区基于自身监管基础采取的不同措施,对我国探索与既有监管基础相适应的做法提供了思路;在原料药及分段生产产物的单独管理方面,美国和日本的经验可为我国原料药登记和关联审评审批制度的拓展提供参考;在监管事权划分方面,欧盟集中程序下EMA 和各成员国药品监管部门的事权,对调整我国国家和省级药品监管事权更具探讨意义。
欧盟、美国、日本等国家和地区的MAH 制度,虽然都较好地处理了持证和生产分离管理等问题,但具体的解决措施做法各有不同。在充分调研借鉴其他国家和地区的成功经验同时,我国也需基于行业状况和监管特点,制定和实施最适合我国国情的MAH 制度,以保障公众用药安全。

来源:中国食品药品监管杂志


