您当前的位置:检测资讯 > 实验管理
嘉峪检测网 2024-10-23 08:18
DNA提取的目的是从细胞或组织中分离并纯化DNA。通常包括细胞裂解、去除蛋白质和其他杂质、以及DNA的沉淀和纯化。
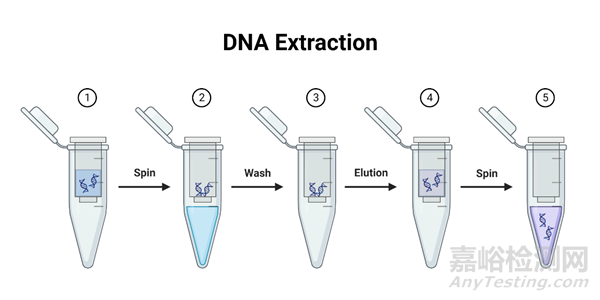
一、离心柱法提取DNA步骤
1)将组织或细胞悬液加入裂解缓冲液,涡旋混匀。裂解缓冲液中含有变性剂(如胍异硫氰酸盐),可以有效地裂解细胞和组织,破坏细胞膜和细胞核膜,同时使核酸与蛋白质分离。
2)将裂解液转移到DNA Spin Column中,离心收集流穿液。这个步骤涉及到将裂解液通过一个特殊的膜过滤,该膜具有特定的孔径,允许小分子如RNA和蛋白质通过,而DNA由于其较大的分子尺寸被截留在膜上。
3)用Washing Buffer洗涤DNA柱膜。Washing buffer中含有乙醇和盐,可以去除杂质和蛋白质,保留DNA在柱膜上。
4)将Elution Buffer加入到柱中,离心收集纯化的DNA。Elution Buffer是低盐缓冲液,能有效洗脱柱膜上结合的DNA,得到纯化的基因组DNA。因为DNA在低盐条件下更容易溶解。离心后,收集纯化的DNA到一个新的收集管中。
5)提取完成后,使用紫外分光光度计测定DNA的浓度和纯度,通过测定260nm和280nm的吸光度比值来评估DNA的质量和蛋白质污染情况。
6)纯化的DNA应妥善存储于-20°C或更低的温度下,以保持其稳定性和防止降解。
二、根据目标基因序列设计PCR引物
在成功提取并纯化了目标基因的DNA后,下一步是设计PCR引物,以便进行目标基因的扩增。合理设计的引物可以确保PCR反应的特异性和效率。设计PCR引物时需考虑以下原则:
1)引物的长度为18-25bp,引物过短可能导致非特异性结合,引物过长可能导致退火效率低。
2)理想的GC含量在45%-55%之间。
3)避免引物内部和引物之间形成二级结构(如发夹结构)和二聚体。
4)引物的3'端应避免过多的G或C,因为这可能导致非特异性扩增。
5)对于基因组DNA,引物不应设计在跨越内含子的区域,以避免扩增非编码区域。
6)引物的3'端不应有过多的连续相同碱基,以减少3'端退化现象。
三、电泳鉴定
1)使用PCR扩增特定的DNA片段。PCR体系:10μL SYBR+ 0.5μL Forward Primer+0.5 μL Reverse Primer+ 1μL 模版DNA+8μL DEPC
2)称取0.3g琼脂糖,加入30mL TAE缓冲液。搅拌均匀后,用锡纸封口。将锥形瓶放入微波炉加热30秒,直至琼脂糖完全溶解,呈现透明无颗粒状态。待溶液冷却至45℃,加入1.5µL核酸染料,轻轻混匀。
3)将溶液倒入提前洗净晾干的制胶板中,厚度为0.3-0.5cm。插入制胶梳,确保胶中无气泡,静置30分钟待胶凝固,小心拔掉梳子。琼脂糖凝胶用于分离不同大小的DNA片段。TAE缓冲液维持电泳过程中适当的pH和离子强度。核酸染料如溴化乙锭嵌入DNA分子中,使其在紫外光下发光,从而可以显影DNA条带。
4)将凝胶连同胶托放入电泳槽正中央,有胶孔的一侧朝向负极。在电泳槽中倒入TAE缓冲液,刚好淹没凝胶。在10µL样品中加入2µL loading buffer,混合后加入胶孔中,加入5µL marker。Loading buffer含有溴酚蓝和二甲苯氰,作为电泳指示剂,显示电泳进程,便于适时终止。甘油增加样品密度,使样品沉降到点样孔中。
5)盖上电泳槽盖,接通电泳仪和电泳槽,设置电压100V-150V,运行30分钟-1小时。电泳结束后,将凝胶放入凝胶成像系统拍摄。在电场作用下,带负电荷的DNA分子从负极向正极迁移。较小的DNA片段迁移得更远。
6)电泳结束后,使用紫外光源观察染料标记的DNA条带,并通过凝胶成像系统拍摄记录结果。
四、实验结果展示

上述是笔者自己做的DNA琼脂糖凝胶电泳图,左侧是Marker(100bp-1500bp),右侧是条带的位置反映了DNA片段的大小。较小的DNA片段移动距离较大,位于下部;较大的DNA片段移动距离较小,位于上部。条带清晰、明确,表明DNA样品纯度高。如果有拖尾或弥散条带,可能表明DNA降解或样品中含有杂质。
五、注意事项
1)在处理多个样本时,使用单独的离心管和离心柱,避免样本之间的交叉污染。
2)设置合适的电压,过高的电压可能导致凝胶过热或DNA条带扭曲。
3)在电泳过程中和电泳结束后,避免凝胶干燥,这会影响DNA条带的清晰度。

来源:实验老司机


