您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-11-27 11:27
当地时间11月21日,欧盟发布《根据MDR 和lVDR逐步推出Eudamed的实施相关实际问题的问答》-经(EU)2024/1860修订,修订了MDR和IVDR法规,涉及逐步推广Eudamed、供应中断或中止情况下的告知义务,以及某些体外诊断医疗器械的过渡性规定
(EU) 2024/1860 对 MDR 和 IVDR 的修订涉及三个主题:
1.(EU)2024/1860 旨在确保高水平的患者安全和公共健康保护,包括在不降低当前质量或安全要求的情况下,降低医疗保健服务顺利运行所需的体外诊断医疗器械(IVD)短缺风险。
为此,根据 IVDR,制造商和公告机构有额外的时间对根据IVDD指令颁发的证书或符合性声明所涵盖的 IVD 进行符合性评估。有关该主题的问答载于另一份文件中。
2. (EU)2024/1860还规定,制造商在中断或停止供应某些医疗器械或IVD之前,必须通知其相关主管当局和卫生机构。如果制造商不直接向医疗机构或医疗保健专业人员供货,则必须通知供应链中的相关经济运营商,然后由其通知医疗机构。这一机制将使主管部门和医疗机构能够考虑采取缓解措施,以确保患者的健康和安全。有关该主题的问题和答案将在另一份文件中列出
3.(EU)2024/1860 还允许逐步推出已完成的欧洲医疗器械数据库(“Eudamed”)中集成的电子系统,而不是将 Eudamed 的强制使用推迟到六个模块中的最后一个模块完成之后。Eudamed 的使用,特别是其经济运营商、器械和证书注册系统的使用,将提高透明度并提供有关欧盟市场上器械的信息,有助于监测器械的供应情况。
本指南就是针对逐步推广 Eudamed 做出的问题和答案。
本指南术语概览
• ACT module: Actor module
• Actor ID: 类似于 SRN 的标识符(结构相同),适用于不在 MDR Article 31 /IVDR Article 28范围内的注册actor。)
• CA: 主管当局
• CI/PS module: 临床调查和性能研究模块
• CECP:临床评价咨询流程
• DA: 指定机构
• NB/CRF module: 公告机构和证书模块
• IVD: IVD器械
• IVDR: Regulation (EU) 2017/746 on in vitro diagnostic medical devices
• MD:医疗器械
• MDCG: Medical Device Coordination Group
• MDR: Regulation (EU) 2017/745 on medical devices
• MfS:审查机制
• MSU module: 市场监管模块
• NB: Notified Body
• NCAR: MDR Article 89 (7) 和 (9) /IVDR Article 84 (7) 和(9) IVDR 条提及的国家主管部门报告
• OJEU: 欧盟官方公报
• PMSV action : 上市后监督(III 类或植入或 CLASS D类监管器械的 PSUR)或与警戒相关的行动(旧的、遗留的、定制的或监管器械的严重事件报告、定期总结报告或FSCA/FSN,或遗留的或监管器械的趋势报告)
• QMS: 质量管理体系
• SS(C)P: 安全性和(临床)性能的总结
• SPP: 系统和程序包
• SPPP: 系统和程序包生产商
• SRN: 单一注册号(根据MDR Article 31 /IVDR Article 28分配给注册参与者)
• UDI/DEV module: UDI/Device module
• UDI-DI: UDI device identifier (pursuant to Article 27(1)(a)(i) MDR/Article 24(1)(a)(i) IVDR)
• VGL module: 上市后监督和警戒模块
Part A- EUDAMED逐步推出(MDR Article 34)
Q1.修订MDR Article 34,使Eudamed逐步推出有何影响?
经(EU) 2024/1860修订的MDR Article 34规定,一旦每个单独的模块经过审计,并且在欧盟官方公报上发布确认模块功能的委员会通知,就可以通过推出单个模块来逐步实施Eudamed。该修正案旨在加速强制使用已确认功能的Eudamed单个模块。
MDR Article 34 的新措辞允许在准备就绪时对单个或多个模块进行独立审计,并在考虑到模块之间相互依存关系的情况下,针对接受审计的模块出具审计报告。
确认一个模块(或一组模块)功能的步骤并没有改变,因为独立审计机构仍然需要核实模块是否符合MDCG和委员会制定的功能规范。一旦委员会在独立审计后核实了这些模块的功能,它将通知并咨询MDCG。随后,委员会将在欧盟官方公报上发布通知,以确认已审计模块的功能。
Part B- EUDAMED强制使用的过渡期(MDR Article 123和IVDR Article 113)
Q2.关于强制使用Eudamed的过渡期,MDR Article 123和IVDR Article 11引入了哪些主要变化?
根据MDR Article 123(3)(d)和IVDR Article 113(3)(e)的新措辞,与Eudamed特定模块相关的义务和要求将在欧盟官方公报发布确认给定模块功能的通知6个月后开始适用。
根据MDR Article 123(3)(d)和IVDR Article 113(3)(e),在与特定Eudamed模块相关的义务和要求成为强制性的日期之前,与警惕性、clinical investigations/性能研究、器械和经济经营者注册以及证书通知相关的相应指令条款和义务适用。这就规定了一个明确的截止日期,即指令的规定(以及相应的国家转运措施)不再适用,与Eudamed相关的规定成为强制性的,从而防止了双重注册问题。
注:(EU) 2024/1860删除了MDR Article 120(8)和IVDR Article 110(8),其中规定,从确认Eudamed功能性的通知发布到其强制用于器械和证书注册的过渡期间,使用Eudamed的器械和证书的注册将被视为符合国家注册要求。
PART C - 每个模块的过渡时期
Eudamed由六个模块组成:
• Actor module (ACT module)
• UDI/Device module (UDI/Device module)
• Notified bodies and certificates module (公告机构和证书模块)
• Market surveillance module (市场监管模块)
• Post-market surveillance and Vigilance module (上市后监督和警戒模块)
• Clinical investigations/performance studies module (临床调查和性能研究模块)
ACTOR MODULE
Q3.什么时候必须使用ACTOR MODULE?
MDR Article 31和IVDR Article 28范围内的经济经营者(制造商、进口商和授权代表)必须注册为行动者,并在适用的情况下,在器械投放市场之前获得单一注册号(SRN)。此外,需要在Actor module 中进行注册,例如,制造商可以在Eudamed中注册器械和警戒报告,或进行任何其他活动。
有关其他需要在Eudamed中注册为参与者的经济经营者或参与者类型的更多细节,请参见Q4。
根据MDR Article 31和IVDR Article 28,在欧盟官方公报公布确认其功能的通知6个月后,Actor module 的注册将成为强制性的。
Q4.是否存在不属于MDR Article 31和IVDR Article 28范围的经济经营者或其他类型的行为者需要在Actor module 中注册?
是的,任何需要在Eudamed中执行动作的actor都需要在Actor module 中注册。这意味着以下经济经营者或其他actor也需要在Actor module 中注册:
- 系统或程序包生产商(SPPP)在将系统或程序包投放市场之前,需要在 ACT 模块中注册并获得一个 Actor ID(类似于 SRN)。
- 专门在市场上销售定制器械的制造商需要在 ACT 模块中注册并获得 Actor ID,然后才能使用 Eudamed 的其他模块,例如报告有关定制器械的严重事故。
- 专门在市场上销售III 类定制植入器械的制造商需要在 ACT 模块中注册,以便 NB 注册根据MDR Article 52(8) 第 2 项颁发的 QMS 证书。
- 临床调查/性能研究的申办者需要在 ACT 模块中注册为行为者,并获得Actor ID,以便能够使用 CI/PS 模块,例如提交临床调查、性能研究或严重不良事件报告的申请。
欲了解更多信息,请参阅委员会关于欧洲医疗器械数据库 (Eudamed) 的实施Regulation (EU) 2021/2078 、MDCG 2021-13 Rev. 1 - “关于制造商、授权代表和进口商以外的行为者在欧盟医疗器械数据库 (EUDAMED) 注册的义务和相关规则的问答(须遵守MDR Article 31和IVDR Article 28 的义务)”,以及关于Actor角色和 Actor ID/SRN的信息图表。
Regulation (EU) 2021/2078
https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/uri=CELEX:32021R2078
MDCG 2021-13 Rev. 1
https://health.ec.europa.eu/system/files/2021-07/md_mdcg_2021-13_q-a-actor_registr_eudamed_en_0.pdf
关于Actor角色和 Actor ID/SRN的信息图表
https://webgate.ec.europa.eu/eudamed-static/infographics/md_actor_roles_srn_en_0.pdf
Q5. 哪些经济运营商无需在 Eudamed 注册?
分销商无需在 Eudamed 注册。但是,分销商可能有义务根据其提供设备的成员国适用的要求在国家层面进行注册。制造商(及其授权代表)、进口商和 SPPP 在强制使用 UDI/DEV 模块后不再向市场投放器械或 SPP 的,无需注册为 Actors。但是,如果需要采取 PMSV 行动,制造商(及其授权代表)应进行注册。
Q6. 我什么时候可以在Actor module 中注册我的组织?
自 2020 年 12 月起,制造商、进口商、授权代表和 SPPP 已经可以在 ACT 模块中注册。强烈建议所有相关经济经营者(不包括分销商)立即在 Actors 模块中注册,以确保在强制使用 Actors 模块之日之前提交注册。
注意:只有当临床调查和性能研究模块成为强制性使用时,才可以注册sponsor。
UDI/DEVICE MODULE
澄清 Eudamed 中 'device/SPP' 注册中 “ 'device/SPP' ”的含义:
在 MDR 和 IVDR 中,“device/设备 ”一词几乎仅指每个单独的设备,即在某个时间点生产并投放市场的每个(销售)单位或单个产品项目。然而,在 Eudamed 中,UDI/DEV 模块中的设备/SPP 注册是指在设备标识符(不包括生产标识符)层面注册设备/SPP。设备/SPP 识别符对于根据条例投放市场的设备/SPP 是 UDI-DI,而对于遗留设备则是 EUDAMED ID 或 UDI-DI。这意味着在 UDI/DEV 模块中只有一个注册,每个设备标识符(UDI-DI 或 Eudamed ID)涵盖所有设备/SPP,但有不同的生产标识符(UDI-PIs),代表批号、序列号、生产日期等。
Q7. 何时强制使用 UDI/DEV 模块?
MDR Article 29 和 IVDR Article 26规定的有关 Eudamed UDI/DEV 模块中器械和系统以及程序包注册的义务和要求将在 OJEU 上发布确认其功能的公告 6 个月后开始适用。
在强制使用日期当日或之后投放市场:
如果在强制使用UDI/Device module的日期或之后,第一个单独(销售)单元的法规器械(定制器械、研究器械和 性能研究器械除外,这些设备不应在UDI/Device module中注册)或具有特定UDI- di的SPP在欧盟市场上投放,则必须在第一个单独单元投放欧盟市场之前在UDI/Device module中进行相应的器械注册。该器械注册涵盖随后以相同的UDI-DI投放市场的所有单个器械。
如在强制使用日期前完成在市场上的出售:
如果在强制使用UDI/Device module的日期之前,遗留器械或法规器械(定制器械、试验器械和性能研究器械除外)的第一个(销售)单元或具有特定UDI-DI的SPP已投放欧盟市场,并且属于同一UDI-DI的其他(销售)单元将在该日期或之后投放市场,UDI/Device module中相应的器械注册必须在欧盟官方公报发布确认UDI/Device module功能的通知后12个月内完成。此器械注册涵盖属于同一UDI-DI的所有单独单元。
例如:如果确认UDI/Device module功能的通知于2025年7月1日在欧盟官方公报上发布,那么MDR Article 29和IVDR Article 26中的UDI/Device module相关要求将于2026年1月1日开始适用。如果器械的单个单位/单个产品在2026年1月1日之前投放市场,而其他单个单位/单个产品也将在该日期之后投放市场,则该器械必须在2026年7月1日之前注册。
最后,MDR Article22(1)-(3)涵盖的spp必须在UDI/Device module中由相关spp注册。在MDR Article22(4)所涵盖的情况下,如果SPP被视为具有自身权利的器械,则需要在UDI/Device module中注册为“器械”
注意:当在UDI/Device module中提交所有必需的器械信息时,制造商已根据第29条MDR 和26条IVDR遵守其关于器械注册的义务。对于某些器械,只有当NB在NB/CRF module中输入相应的产品证书信息后,UDI和器械数据才会对公众可见。
Q8. 哪些器械不需要在UDI/Device module中注册?
当UDI/Device module成为强制性时,单个(销售)单元不再投放市场的遗留和监管器械不需要注册,除非发生PMSV action。
此外,如果“‘the same device/相同器械”已经注册为法规器械,则不需要注册遗留器械。在这种情况下,“相同器械”是指法规器械和遗留器械具有相同的标识,如UDI-DI,和/或目录/参考编号和/或商品名称,这些标识来自共同的特征。见Q14中的例外。
注意:对法规器械进行更改的器械,将导致分配新的UDI-DI,将不被视为“‘the same device/相同器械”。
MDCG 2021-25 Rev. 1和MDCG 2022-8中描述的“旧”器械不能在UDI/Device module中注册。如果器械是严重事故报告(MIR)或现场安全性纠正措施(FSCA)的主题,则、制造商将需要提供有限的器械数据集,以便在上市后监督和警戒模块中提交相关报告。
定制的器械也不能在UDI/Device module中注册。如果定制器械是MIR或FSCA的主题,制造商将需要提供有限的器械数据集,以便在 VGL module中提交相关报告。
Q9. 我何时可以开始在 UDI/DEV 模块中注册器械/SPP?
UDI/DEV 模块自 2021 年 10 月起可供自愿使用。设备/SPP 自该日起即可注册。强烈建议尽快在 UDI/DEV 模块中注册设备和 SPP,而不要等到其强制使用开始。
注:在 UDI/DEV 模块强制使用之前,可能适用国家注册要求。一旦 UDI/DEV 模块开始强制使用,在国家系统和 Eudamed 中都进行注册的情况下,已在 UDI/DEV 模块中注册的设备/SPP 将成为法定注册。
Notified bodies and certificates module (公告机构和证书模块)
Q10. 何时强制使用公告机构和证书(NB/CRF)模块?
与 Eudamed NB/CRF 模块相关的义务和要求将在 OJEU 发布确认其功能的公告 6 个月后开始适用。这意味着,在 NB/CRF 模块强制使用后签发的任何证书都必须在 NB/CRF 模块中注册。强制使用 NB/CRF 模块后发布的与法规有关的更新和决定
在强制使用 NB/CRF 模块之前签发的证书,必须在 NB/CRF 模块中登记。
下列规定与 NB 有关:
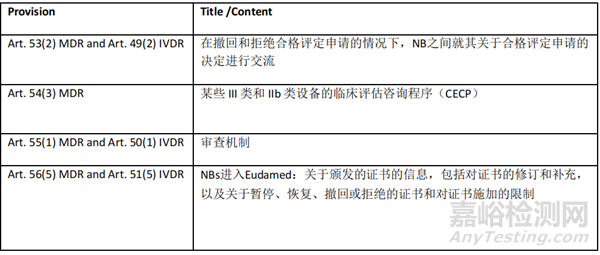
有关DAs的规定如下:

对于在强制使用 NB/CRF 模块之前根据 MDR/IVDR 签发的证书,NB 必须在 OJEU 上发布确认 NB/CRF 模块功能的通知后 18 个月内在 Eudamed 注册相关信息。这仅适用于需要或已在 UDI/DEV 模块中注册的监管设备。此外,只有最新的证书版本以及与该证书版本相关的最新 NB 决定(如适用)才应在 Eudamed 中注册。
注:根据 MDR Articles 40(2), 42(10) and 43(2) 以及 IVDArticles 36(2), 38(10) and 39(2) (公布联合评估的国家专家、NANDO 检索的 NB 名单以及相关通知),委员会将在 Eudamed 中管理相关信息。
关于 CECP(Article 54 MDR)和审查机制(Article 55(1) MDR/Article 50(1) IVDR))的说明
在 NB/CRF 模块成为强制性使用后,当证书在 NB/CRF 模块中注册时,在 Eudamed 审查机制 (MfS) 下提供信息的义务将适用。
在强制使用 NB/CRF 模块之前,可在不具备 CECP 和 MfS 功能的情况下在 Eudamed 注册证书。对于这些证书,在强制使用后,将有可能说明 CECP 以及 MfS 是在 Eudamed 之外进行的。如果在强制使用 NB/CRF 模块之前,NB 没有在 Eudamed 之外根据 MfS 进行通知,则必须在 Eudamed 注册证书时进行通知,即使 CECP 注册是在 Eudamed 之外进行的。
关于 SS(C)P 的说明
自相关证书在 Eudamed 注册之时起,NB 即有义务根据 MDR Articles 32(1) 和 IVDR 29(1) 上传 SS(C)P。
Q11. NB何时可以开始使用 NB/CRF 模块?
NB/CRF 模块自 2021 年 10 月起可供自愿使用。通知机构已可在 Eudamed 注册证书和 SS(C)P。
如果使用 NB/CRF 模块注册证书信息,则与首次注册证书相关的所有后续更新和决定(如撤销、暂停、恢复)都必须在 Eudamed 中注册(这也适用于自愿使用期间)。
强烈建议尽快在 NB/CRF 模块中注册证书,而不要等到开始强制使用时才注册
上市后监督和警戒模块
Q12. 何时强制要求使用上市后监督和警戒 (VGL) 模块?
与 Eudamed VGL 模块相关的义务和要求将在 OJEU 发布确认其功能的公告 6 个月后开始适用。
以下规定适用于 CA、NB、制造商和/或授权代表:
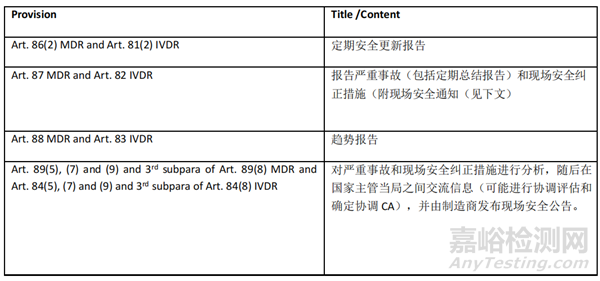
注:Articles 90 MDR and 85 IVDR(警戒数据分析)将在 VGL 模块第一个强制版本推出后实施。
Q13. 何时可以开始使用 VGL 模块?
VGL 模块目前还不能自愿使用。它将在成为强制版本后发布,届时必须使用。因此,经济运营商需要继续使用 MDCG 2021-1 Rev. 1 和 MDCG 2022-12 中解释的国家流程,以遵守 MDR/IVDR 警惕规定。制造商和授权代表在 Eudamed 中提交所需的 PSUR(请参阅缩写部分)和警戒报告,以及 CA 提交 NCAR,将在 VGL 模块成为强制使用时开始。
注:对于在没有 Eudamed 的情况下根据国家程序启动的警戒报告,如果在 VGL 模块强制使用时仍未完成,则应在 Eudamed 中完成后续行动。
这并不意味着对警戒报告进行追溯登记,只有从强制使用 VGL 模块时开始的行动才应在 Eudamed 中进行。
Example: 2026 年 4 月 1 日按照国家流程报告了一起与设备相关的严重事故。如果 VGL 模块于 2026 年 7 月 1 日成为强制性模块,且严重事故报告 (MIR) 的后续版本于 2026 年 8 月 1 日可用,则应在 Eudamed 中报告此版本的 MIR。
Q14:哪些设备只有在发生 PMSV 行动时才需要在 UDI/DEV 模块中注册?
自 UDI/DEV 模块成为强制性模块之日起,没有单件(销售)投放市场的管制设备和传统设备,只有当制造商必须在 VGL 模块中执行任何 PMSV 操作时,才需要在 UDI/DEV 模块中注册(另见 Q8)。为此,制造商和授权代表(如适用)必须首先在 ACT 模块中注册为 Actor。
Example: 某监管设备于 2021 年 5 月至 2025 年 11 月期间投放市场。如果 UDI/DEV 模块于 2026 年 1 月 1 日成为强制性模块,则该设备无需在 Eudamed 中注册。如果 VGL 模块于 2026 年 7 月 1 日成为强制性模块,并且在 2026 年 9 月发生了与该设备相关的严重事故,则必须在 UDI/DEV 模块中注册该设备,以便在 VGL 模块中报告该严重事故。
如果要在 Eudamed 中报告的警戒行动(如严重事故报告、FSCA/FSN 或趋势报告)涉及遗留器械而非 “同一 ”规管设备,则遗留器械必须例外地在 UDI/DEV 模块中注册,并在 VGL 模块中输入 PMSV 行动参考。这并不影响以下原则:如果 “同一设备 ”已注册为监管设备,则遗留器械无需注册(请参阅问题 8,了解何为 “同一设备”)。
Market surveillance module (市场监管模块)
Q15. 市场监督 (MSU) 模块何时成为强制性模块?
与 Eudamed MSU 模块相关的义务和要求将在 OJEU 发布确认其功能的公告 6 个月后开始适用。
注:MSU 模块仅限主管当局和 NB 查看(如适用),且该模块的数据仅由主管当局管理。
Q16. 主管当局何时可以开始使用 MSU 模块?
MSU 模块并非自愿使用。它将在成为强制性标准时发布,届时主管当局必须使用。
临床调查和性能研究模块
Q17. 临床调查/绩效研究(CI/PS)模块何时成为强制性模块?
与 Eudamed CI/PS 模块和申办者在 ACT 模块中注册有关的义务和要求将在 OJEU 发布确认 CI/PS 模块功能的通知 6 个月后开始适用。
Q18. Articles 78(14) MDR and 74(14) IVDR 中提及的协调评估何时开始适用?
协调评估程序将在 OJEU 发布确认 CI/PS 模块功能的通知 6 个月后开始适用于同意适用该程序的成员国(“自愿 ”阶段)。在OJEU发布确认 CI/PS 模块功能的公告 5 年后,当申办者提交单一申请时,协调评估将成为所有成员国的强制性程序
Q19. 在没有 Eudamed 的情况下,协调评估程序是否可行?
Article 78 MDR and Article 74 IVDR 中提及的临床研究和性能研究的协调评估程序基于 Eudamed 的 CI/PS 模块的使用。然而,在 CI/PS 模块可用之前,成员国和申办者可根据 MDCG 提供的指导,同意安排使用替代方法进行协调评估。
Q20. 什么时候可以开始使用 CI/PS 模块?
CI/PS 模块不是供自愿使用的,它将在成为强制性模块时发布,届时必须使用。

来源:北京倍力医疗技术服务有


