您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-12-02 09:45
对任何一种药物来讲,临床剂量的选择和优化都是重要课题。对于肿瘤药物,传统思路是在临床Ⅰ期阶段探索最大耐受剂量(MTD),之后在Ⅱ期和Ⅲ期临床中确认该剂量下的有效性。这种思路源自“more is better”的假设,底层逻辑是剂量越高,药效越好。对于细胞毒类药物,这种思路或许是对的,毕竟细胞毒类药物量效关系比较陡峭,升高剂量,药效影响可能会比较大。但对于靶向化药或抗体,治疗范围非常宽,低于MTD的剂量可能与MTD活性类似,或者有些药物因安全性好无法达到MTD,甚至观察不到剂量限制性毒性(DLTs),就不太适合用MTD进行剂量选择,应该寻找最佳生物学剂量(optimal biological dose, OBD)。另外,剂量优化不理想会产生诸多隐患,比如药物研发失败,临床期间多次进行剂量调整,上市后依然需要进行剂量方面的研究。这些操作会非常耗时、耗力,增加成本。
相较于其他治疗领域,肿瘤药物有其特殊性。首先大部分肿瘤会威胁生命,药物开发速度与患者受益之间的关系更密切。其次,肿瘤药物开发的主要障碍是缺少有效性。出现这一问题很重要的一个原因是患者给予过高剂量,短期剂量爬升阶段未出现毒性,但长期给药导致的毒性使很多患者不得不降低剂量、给药中断,从而影响有效性。
2021年,FDA肿瘤卓越中心(Oncology Center of Excellence)启动了一项名为Optimus的项目,旨在改革肿瘤药物研发中的剂量优化和剂量选择模式。2024年8月,FDA发布了《Optimizing the Dosage of Human Prescription Drugs and Biological Products for the Treatment of Oncologic Diseases》指导原则,帮助进行肿瘤药物剂量优化。
FDA认为剂量选择应该充分考虑非临床和临床研究数据(比如PK、PD、安全性、耐受性、给药便利性和活性),并对剂量或暴露量-效应关系进行分析。如果不能对选定的剂量给出充分且合理的解释,FDA原则上是不接受的。FDA鼓励申办方期临床阶段就剂量优化方案与FDA召开正式会议。FDA对如何进行剂量优化也给出了一些建议。
首先,临床PK、PD和药物基因组学方面。剂量探索试验中,PK样本收集和分析需要充分,以完整表征产品的PK特征,如线性、吸收、分布、消除等,并支持群体PK和暴露量-效应关系分析。如果条件允许,建议收集PD和药物基因组学样本,用于药物代谢酶或转运体基因、肿瘤基因或靶蛋白表达等分析。FDA建议群体PK分析宜尽早启动,界定体重、年龄、性别、种族、器官损伤等特定人群的影响。剂量/暴露量与效应之间的关系也应尽早开展。结合PK、PD、群体PK和剂量-反应关系、暴露量-反应关系,及其他数据如安全性、耐受性、给药便利性和活性等数据,选定合理剂量。也可以使用模型支持剂量选择。
然后再看下不同剂量对比的试验设计。FDA认为同类药物或同作用机制的药物临床数据可以作为参考。模型对鉴定和选择拟比较的剂量是有帮助的。另外,FDA推荐开展随机、平行方案设计进行剂量间对比。样本量方面,不需要证明剂量间的统计学优效或非劣效,但要有足够的样本量测定每个剂量的安全性和抗肿瘤活性。抗肿瘤活性的终点既可以是ORR、PFS,也可以包括其他组织、血液或基于成像的终点。多剂量对比设计既可以置于关键注册临床试验之前,也可以作为关键注册临床的一部分,即增加剂量组。
以下是《Design Strategy and Consideration for Oncology Dose-Optimization: An Industry Perspective》一文给出的5个剂量优化时需重点考虑的因素,本文是Regeneron、MD Anderson、Novartis、Bayer、Seagen、BeiGene、AstraZeneca等一些大药企联合撰写的,有兴趣可以翻看原文。
毒性和有效性终点
毒性和有效性的终点对于评估潜在的获益和风险是很重要的。比如常见的毒理终点DLT,通常代表治疗周期内的3级及以上AE。不过,仅评估DLT是远远不够的,需要更全面表征低等级或蓄积毒性。低等级毒性可以考虑用总毒性负荷(total toxicity burden, TTB)评估,即每种类型和等级的AE均赋予一个评分,最后对所有AE进行汇总。也可以考虑用等效毒性评分方法(equivalent toxicity score, ETS),即每种低等级毒性折算成DLT的比例,比如2级毒性计为0.5 DLT。对于蓄积毒性,可以用剂量耐受比例(dose tolerability rate, DTR)进行评估,即多次给药后,给药终止/剂量降低/给药中断的比例等。
比较熟知的有效性终点包括客观缓解率(ORR)、无进展生存期(PFS)。对于早期临床ORR、DOR(缓解持续时间)、PD biomarker(如ctDNA)更为常用。采用缓解相关的指标会面临一个挑战,即有些药物或患者需要多次给药后才能有效,这就不支持实时进行剂量方面的决策。所以,基于PD的终点或靶点受体占有率,这类短期替代终点更为快捷、方便、实用。长期终点可以在试验结束后,再进行总体分析。如果不同角度的证据如PK、PD和有效性等均指向同一个剂量,当然更具说服力。比如PD-1抗体nivolumab采用受体占位、PK参数和肿瘤缩小共同辅助进行剂量选择。
风险-获益权衡标准
剂量优化的目的是基于风险-获益的权衡(benefit-risk trade-off, BRTO)获得一个理想剂量。不过,BRTO在实际操作中用的还不多,大家普遍还是沿用之前的简单路径,即先找到几个可以耐受的剂量,选择其中一个药效最好的最大耐受剂量作为理想剂量。举个简单的例子,比如剂量二高于剂量一,剂量一和剂量二药效终点表现类似,但剂量一毒性发生率低,传统路径可能选择剂量二,因为更接近MTD,而基于BRTO的思路,应该选择剂量一。
还有一种更为灵活和多见的方法是医生对每个患者每个剂量下的有效性和安全性终点进行打分。常见的有4种结果(有效性、毒性)=(yes, no),(yes, yes),(no, no),(no, yes)。最希望出现的结果肯定是(yes, no),赋100分;最不希望看到的是(no, yes),赋0分;(yes, yes)和(no, no)则分别赋60和40分。最后根据每个剂量所有患者的平均分进行剂量选择。
不过,以上都是些简单示例,BRTO是一个多维度,涉及多个终点的路径,很难通过单一的标准衡量所有终点的获益-风险。
统计模型
统计模型主要用于剂量/暴露量和效应(如毒性、有效性)之间的关系评估。常用的两种模型策略分别是model-based和model-assisted。EffTox和late-onset(LO)-EffTox设计就是早期临床用于剂量优化的model-based的案例,通常需要制定参数或非参数剂量-反应关系曲线(如对数或Emax模型)。Model-assisted涉及的模型则更为简单,不需要实时模型评估,也不需要搭建剂量-毒性或剂量-有效性曲线。有点类似于3+3设计,会在试验启动前有一个决策表。
统计这部分有点超出个人专业,不再展开,大家可以翻看下原文。
临床设计架构
常见的两种设计思路分别是两阶段优化策略(two-stage optimization strategy)和有效性整合优化策略(efficacy-integrated optimization strategy)。
两阶段优化策略比较常见的是两阶段无缝衔接的设计,如Ⅰ/Ⅱ、Ⅱ/Ⅲ期合并。
Ⅰ/Ⅰb期剂量爬升设计探索最佳生物学剂量
传统MTD路径其实是依赖单一毒理学指标进行剂量设计。过去20多年,Ⅰ期爬坡设计已经出现很多新的方法。大部分方法具备既可升高,又可回调剂量的特点,且同时考察有效性和安全性两个变量,甚至还包括biomarkers、PD终点,以期发现既安全又有效的理想剂量。不过,这类设计有些局限性。首先,有效性通常比安全性需要更久的随访时间,剂量爬升期间操作起来会有困难。其次,首次临床试验入组患者通常是末线,一般不限瘤种,患者间变异较大。而且,有效性受肿瘤类型、患者异质性影响较安全性更大。这也是为什么Ⅰ期临床更多目的是考察耐受性、安全性,而不是有效性的原因。另外,大部分新型爬坡方案需要用到复杂的Bayesian模型,需要统计学家参与,涉及到的参数比较多,操作起来要求具备一定的专业性。所以,在Ⅰ/Ⅰb期阶段,同时结合有效性和安全性数据进行最佳剂量探索是要面临一定挑战的。
不过,挑战和问题就是用来克服的。大部分药企还是倾向于在早期临床阶段就开始剂量优化的工作。尤其肿瘤属于威胁生命的疾病,入组早期临床的患者也能有更大机会在最优剂量下用药。
无缝临床Ⅰ/Ⅱ期剂量探索设计
Huang等人在Some Statistical Considerations in the Clinical Development of Cancer Immunotherapies一文中提出一种相对务实的方法,分三步。第一步先用Bayesian模型评价安全性。第二步对PK、PD biomarkers、暴露量-效应关系及初步药效进行分析。第三步在某一目标人群中采用随机分组的形式扩展1-3个剂量确认有效性。
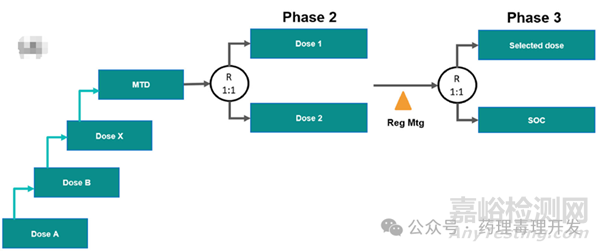
无缝临床Ⅱ/Ⅲ期剂量探索设计
Ⅱ/Ⅲ期无缝连接更多是为了节约时间。示意图如下图所示,通过一定样本量的Ⅱ期随机剂量探索试验完成剂量优化
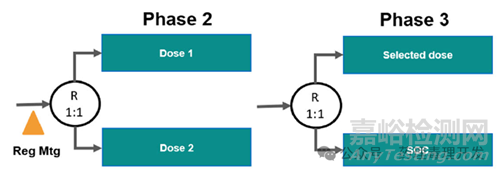
有效性整合策略也叫fully sequential phase Ⅰ/Ⅱ designs,旨在药物临床开发初期就开始优化剂量,在剂量爬升/降低时,同时考虑有效性和安全性。与前文中Ⅰ/Ⅱ两阶段策略相比,有效性整合策略没有将剂量探索从剂量优化阶段中剥离出去,而是持续入组,根据最新更新的数据,将新入患者分到某一剂量,直至达到最大样本量或满足其它终止规则。
可接受和终止规则
这一规则主要用于保护患者免于暴露在无效剂量或毒性剂量。可接受的剂量指的是某一剂量满足预设的有效性、安全性标准。如果试验中发现已经没有可接受剂量,试验可以终止或者视情况增加一个中间剂量。
案例
为了探索sitravatinib与nivolumab在肾细胞癌中联用的有效性和安全性,进行了一种基于有效性整合的LO-EffTox trade-off Ⅰ/Ⅱ期设计。主要终点有两个,一个是12周内的DLT,作为安全性终点。一个是6周的早期有效性终点。主要探索sitravatinib的最优剂量,nivolumab为固定剂量。Sitravatinib给予80mg和120mg两个剂量。两个剂量的风险-获益权衡标准的评分是一样的,所以要借助于额外的标准。Bayesian后验概率,120mg较80mg的ORR更高、PFS更长。与80mg相比,120mg具备相似的安全性表现,但患者抑郁发生率更低、生活质量更高。故,120mg被认定为最优剂量。
再看一个两阶段优化策略的案例。Belantamab mafodotin是一个BCMA靶点ADC,设计的是Ⅰ/Ⅱ期两阶段临床策略。DREAMM-1首次人体试验,先进行爬坡,0.03-4.6mg/kg,IV,Q3W,未达到MTD。选择3.4mg/kg进行扩组,虽然看到活性,但71%的患者中断给药,66%患者出现降低剂量情况。为改善耐受性,开展了2.5和3.4mg/kg两个剂量对比的DREAMM-2试验,患者被随机分组接受这两个剂量给药。样本量根据预设的33%或更高的有效性进行计算。结果显示,2.5mg/kg(97人)和3.4mg/kg(99人)的ORR分别为31%和34%,旗鼓相当。不过,2.5mg/kg在致死性AE,严重AE,剂量中断和剂量降低方面表现更好。最终,选定2.5mg/kg作为最优剂量进入下个阶段。

来源:药理毒理开发


