您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-12-12 19:17
一、基本概况
1、自然环境
美国位于北美洲中部,领土还包括北美洲西北部的阿拉斯加和太平洋中部的夏威夷群岛,北与加拿大接壤,南靠墨西哥湾,西临太平洋,东濒大西洋。国土面积937万平方公里。本土东西长4500公里,南北宽2700公里,海岸线长2.27万公里。
2、人口和行政区划
1、人口分布
根据美国人口调查局最新统计,2022年12月美国人口为3.33亿。人口最多的前五个州是:加利福尼亚州(3903万)、得克萨斯州(3003万)、佛罗里达州(2224万)、纽约州(1968万)、宾夕法尼亚州(1297万)。
2、行政区划
美国共分50个州和1个特区(哥伦比亚特区,首都华盛顿所在地),有3143个县。联邦领地包括波多黎各和北马里亚纳;海外领地包括关岛、美属萨摩亚、美属维尔京群岛等。首都华盛顿哥伦比亚特区,人口约68万。主要经济中心城市包括:纽约、洛杉矶、芝加哥、底特律、亚特兰大、波士顿、西雅图等。
3、2024年出口概况
2024年1-5月,中国向美国出口医疗器械总计约273.0亿人民币,同比增长约6.41%;2024年1-6月,美国新增批准医疗器械产品总计2,953款,其中859款由中国企业注册;出口欧美国家的主力省份如图所示。

二、美国医疗器械监管机构和法规要求
美国医疗器械监管机构为食品药品监督管理局(Food and Drug Administration, 简称FDA)。
在美国销售的医疗器械需遵守联邦食品、药品和化妆品法案(the Federal Food, Drug, and Cosmetic Act, 简称FD&C法案)监管要求,以及联邦法规21 CFR(Title 21-Code of Federal Regulations)第1-58部分和第800-1299部分的相关规定。
三、医疗器械定义
医疗器械涵盖的范围广泛,从压舌板等简单或低端耗材,到可编程心脏起搏器和闭环人工胰腺系统等复杂或高端设备;还包括体外诊断(IVD)产品,例如试剂、测试套件和血糖仪。此外,某些用于医疗用途或声称具有医疗功能的辐射或发射电子产品也被视为医疗器械,如诊断超声产品、X射线机和医疗激光设备。
● 根据FD&C法案第201(h)(1)条的规定,医疗器械是指:
仪器、仪表、器具、设备、装置、植入物、体外诊断产品或其他类似或相关的物品,包括以下产品或附件:
(1) 收录在国家处方集、美国药典或其补充性文件;
(2) 拟用于诊断疾病或其他病症,或用于治愈、减轻、治疗或预防人类或其他动物的疾病;或
(3) 旨在影响人体或其他动物身体的结构或任何功能,并且不能通过人体或其他动物体内或体外的化学作用达到其主要预期目的,并且不依赖于代谢来实现其主要预期目的。术语“设备”不包括根据第520(o)条排除的软件功能。
四、医疗器械产品分类
医疗器械根据其风险程度分为三类(I类、II类或III类)。随着器械类别从I类到II类再到III类,监管控制强度也逐步增加,I类器械受最少的监管控制,而III类器械则受到最严格的监管控制。器械类别、监管控制和提交类型总结如下表所示:

可以通过以下三种方法确定产品分类
1、检索产品分类数据库
您可以通过检索FDA产品分类数据库 FDA Product Classification Database,确定是否有适用于您的产品的现有产品分类:
◆ 使用“快速搜索”功能按关键词进行搜索。请注意,您可能需要使用多 种描述产品的关键词进行多次搜索。
◆ 使用“高级搜索”功能按产品代码、法规编号或器械类别进行搜索。
2、检索类似器械
如果您确定了在美国合法销售的类似器械,您可以查找FDA发出的允许市场授权的信函或命令。该信函或命令中关于类似器械类型的信息可能有助于您确定您器械的分类。
FDA允许市场授权的决策是公开信息,您可以通过使用“快速搜索”或“高级搜索”功能在以下数据库中进行搜索来查找这些信息。
◆ 上市前批准 (PMA)数据库——大多数III类(高风险)器械在合法销售前需要获得上市前批准(PMA)。该数据库包含获得上市前批准的器械,包括批准命令、安全性和有效性摘要,以及批准器械的标签(包括原始PMA和跟踪补充)。
◆ 上市前通知 510(k)数据库——大多数II类(中等风险)器械在合法销售前需要获得FDA的510(k)许可。该数据库包含可公开的510(k)信息。
◆ De Novo数据库——De Novo为低至中等风险的新型器械提供了一种可能的分类途径。该数据库包含De Novo分类和公开摘要。
◆ 人道主义器械豁免 (HDE)数据库—— HDE提供了一种可能的途径,使可能有助于治疗罕见疾病或病症的医疗器械进入市场。该数据库包含获得HDE批准的器械,以及批准命令、安全性和可能受益的摘要及批准器械的标签。
注意:大多数I类和部分II类器械可能未列入上述四个数据库,因为它们被豁免,无需在上市前经过FDA审核。
此情况下,您可以通过查看器械的列名信息来检索合法销售的器械的产品分类。器械列名信息可以通过使用FDA的“ Establishment Registration and Device Listing database”数据库的快速搜索或高级搜索功能进行查询。
3、产品分类申请
如果想从FDA获得正式的产品分类,可以考虑递交513(g)申请,FDA在收到任何一份513(g)书面申请后的60天内,会提供该医疗器械分类的书面声明以及此设备基于该法案适用的要求,具体的流程见官方指南文件FDA and Industry Procedures for Section 513(g) Requests for Information under the Federal Food, Drug, and Cosmetic Act | FDA
额外关注:器械软件功能
FDA对器械软件功能采用与其他医疗器械相同的基于风险的方法,以确保其安全性和有效性。指导文件《Policy for Device Software Functions and Mobile Medical Applications》提供了FDA可能如何监管某些器械软件功能的示例。该指导文件还提供了以下软件功能的示例:
◆ 非医疗器械
◆ 是医疗器械,但FDA计划行使监管裁量权的
◆ 是医疗器械并且是FDA监管重点
五、美国医疗器械上市步骤流程
1、上市流程图
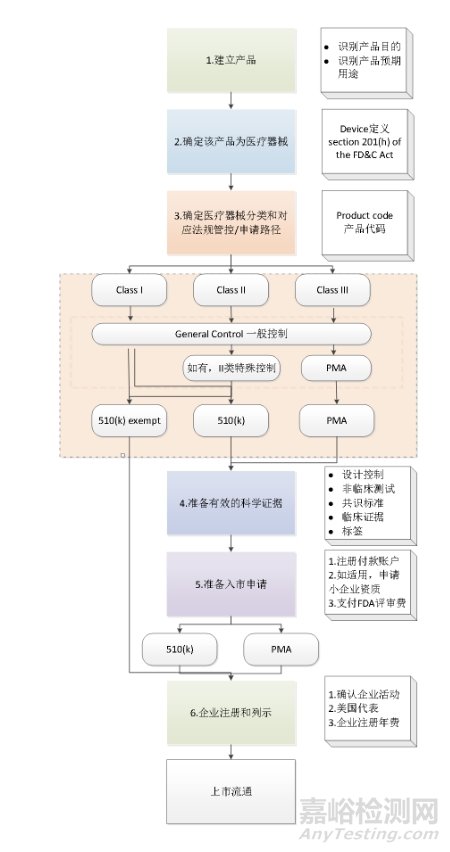
2、注册提交技术文件
1、510(k)文档(以医疗器械产品举例)
① Submission Type
② Cover Letter/Letters of Reference
③ Applicant Information
④ Pre-Submission Correspondence & Previous Regulator Interaction
⑤ Consensus Standards
⑥ Device Description
⑦ Proposed Indications for Use(Form FDA 3881)
⑧ Classification
⑨ Predicates and Substantial Equivalence
⑩ Design/Special Controls, Risks to Health, and Mitigation Measures
⑪ Labeling
⑫ Reprocessing
⑬ Sterility
⑭ Shelf Life
⑮ Biocompatibility
⑯ Software/Firmware
⑰ Cybersecurity/Interoperability
⑱ Electromagnetic Compatibility(EMC), Electrical, Mechanical, Wireless and Thermal Safety
⑲ Performance Testing
⑳ References
㉑ Administrative Documentation
㉒ Amendment/Additional Information(AI) Response
2、PMA文档

3、注册周期及费用
注册周期:按历史数据统计,首次注册周期510(k)约为120个自然日,PMA约为300个自然日。
注册费用:FDA每年8月1日公布下一个财政年收费标准,当年10月1日∼下一年9月30日为一个财政年。
2025财政年收费标准如下:
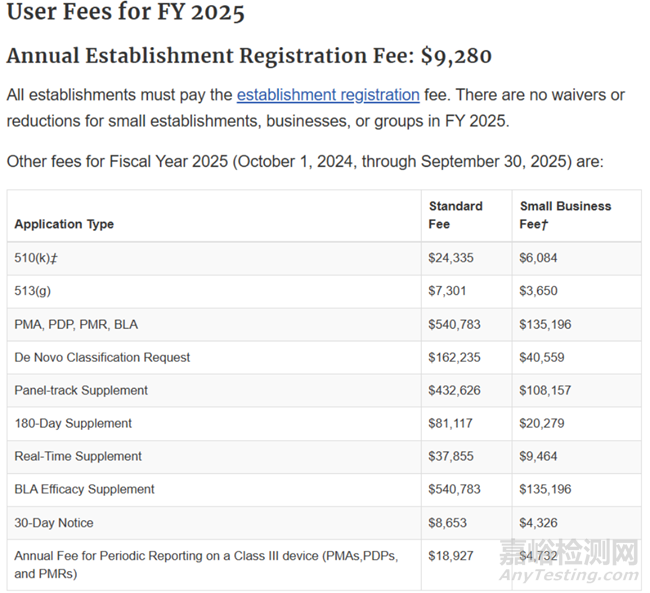
注:企业注册年费缴纳时间:10月1日∼12月31日,不可享受小微企业优惠。
经过Center for Devices and Radiological Health (CDRH) 认定的小微企业(企业及其所有分公司的最近1年税年总营业额小于1亿美金)可以享受费用优惠政策,最近FDA更新的2025财政年510(K)、513(g)、PMA、De Novo申请的标准收费和小微企业收费情况见上表所列,官网:Medical Device User Fee Amendments (MDUFA) | FDA。
4、eSTAR
eSTAR (Electronic Submission Template And Resource)是一个交互式PDF表单,引导申请人完成准备全面医疗器械提交的过程。
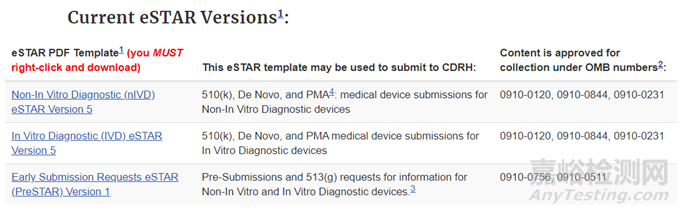

具体eSTAR各子目录文件要求详见官网信息。
从2023年10月1日开始,所有产品的510(K)申请,除非是豁免510(K)的情形,否则强制要求通过CDRH portal或CBER的ESG (FDA’s Electronic Submission Gateway)以eSTAR方式进行递交。De Novo申请、Pre-Sub申请、特定PMA和补充类型申请可以选择自愿通过CDRH portal以eSTAR方式进行递交,或选择以eSTAR方式邮寄FDA。513(g)申请可以选择自愿通过CDRH portal或ESG以eSTAR方式进行递交,或选择以eSTAR方式邮寄FDA。
递交前需先创建递交关口账号,CDRH Portal创建账号,ESG创建账号。
5、现场审核要求
510(k)申请过程无现场审核环节,上市后FDA根据产品投诉或不良事件、出口量、海关抽查情况和审核惯例(Ⅰ类大概每4年左右被抽查一次,Ⅱ类及Ⅲ类大概每2年左右被抽查一次)进行GMP (QSR820)质量体系现场审核。PMA申请过程设有GMP (QSR820)质量体系现场审核环节,如审核通过,将授予GMP Clearance。FDA工作人员基于QSR 820、QSIT质量体系检查指南进行现场审核。
6、企业注册及产品列名
涉及预期在美国商业流通的医疗器械的生产和分销的企业(如制造商、进口商)需要向FDA进行年度注册,完成注册之后可获得注册编号(Registration Number)以及所有者/经营者编号(Owner/Operator Number)。
针对企业所进行的活动类型,例如生产商,一级分销商,规格开发商,合约制造商,合约灭菌商,境外出口商,再生产商和再贴标商、再包装商,一次性使用器械再处理商等不同角色,有相应的企业注册及列示要求,详见FDA官网 Who Must Register, List and Pay the Fee | FDA。
在企业开始活动或将器械投入商业分销后30天内提交企业注册和/或产品列示信息。外国企业在向美国出口产品前必须注册,美国本土进口商在进口产品前必须进行企业注册。企业注册为年度注册,即使信息没有变化,企业注册信息必须在每年10月1日∼12月31日期间提交。
工厂注册及产品列示的具体操作流程见官网How to Register and List | FDA。
7、UDI要求
FDA已全面实施UDI赋码要求,其中Ⅰ类赋码到DI即可,其他类别需赋码到DI+PI。此外,产品投放美国市场前,需将UDI - DI (Ⅰ类)或UDI - DI+PI flag (Ⅱ类及Ⅲ类)上传GUDID数据库。
8、其他注意事项或特别提醒
QMSR (Quality Management System Requirement)已于2024年02月02日发布,两年过渡期,即2026年02月02日起GMP质量体系现场审核将按QMSR进行。
从2025年10月1日开始,De Novo申请将强制要求通过CDRH portal以eSTAR方式进行递交。

来源:广东医疗器械学会


