您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-12-26 09:23
谈到我们在医疗器械安全设计方面的初衷与实践之间的差距。一般来说,我们都会尽最大努力开发和推出被监管机构认定为安全有效的器械。然而,频繁的召回和不良事件报告表明,我们整个行业在安全设计方面仍然举步维艰。我们似乎陷入了一个永无止境的循环,即在产品上市后阶段救火。
一个潜在的原因是风险管理与设计控制之间的脱节。这是否会影响您通过设计最大限度地提高安全性的能力?
设计控制仍然是绝大多数医疗技术公司面临的难题,这已不是什么秘密。根据FDA最近的检查数据,在过去5年中,与设计控制相关的检查结果平均占总检查结果的13%。
如下图1所示,在2019年1月至2024年9月期间结束的检查中,FDA共向医疗器械制造商发出了1117条意见。按百分比计算,设计验证、设计变更、一般控制、设计历史档案和设计验证方面的缺陷占这些检查意见的近80%。

Figure 1: FDA inspectional observations for Design Controls on a percentage basis by subclauses of 21 CFR 820.30. Data source: FDA.
仔细研究21 CFR 820.30(g)条中设计验证方面的首要问题,就会发现风险分析方面的缺陷占这一类338条意见的45%。
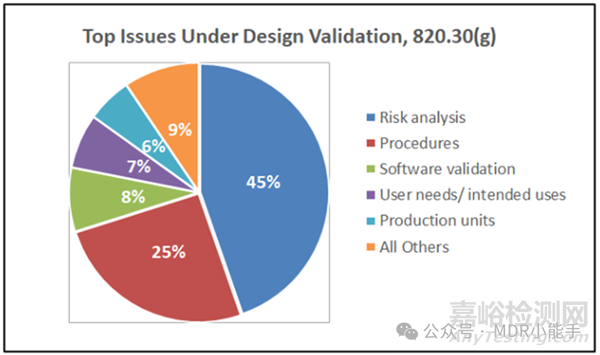
Figure 2: Distribution of top issues related to design validation requirements per 21 CFR 820.30(g). Data source: FDA.
当然,风险管理过程中的漏洞会导致质量管理系统中其他过程(包括CAPA)出现问题。然而,现行的质量体系(21 CFR 820)仅在一个子条款中提及风险分析,即21 CFR 820.30(g)中的设计验证。
现在,FDA已经发布了关于QMSR的最终规定,预计将于2026年2月开始实施,现在是时候对风险管理与设计控制之间可能存在的脱节进行严格审查了。
这是因为ISO 13485:2016是QMSR的基础。它有明确的基于风险的要求,尤其是对设计和开发过程的要求。例如,第7条要求:
The organization shall document one or more processes for risk management in product realization. Records of risk management activities shall be maintained (see 4.2.5).
第7.3.3和7.3.9条对设计和开发过程的基于风险的要求如下:
These (design) inputs shall include - c) applicable output(s) of risk management;
The review of design and development changes shall include evaluation of the effect of the changes on constituent parts and product in process or already delivered, inputs or outputs of risk management and product realization processes.
因此,ISO 13485要求将风险管理活动与设计和开发紧密结合起来。因此,按照现行QS法规的要求,仅关注风险分析已远远不够。这就要求我们大大转变观念,采用更周到、更系统的方法来建立坚实的风险管理能力。
风险管理与设计控制之间潜在脱节的5个例子
根据最近的FDA警告信,您可以考虑检查风险管理与设计控制之间的潜在脱节:
脱节#1:风险评估中遗漏危害
脱节#2:并购后未能更新风险分析
脱节#3:投诉数据分析不充分
脱节#4:危害严重程度的定义不明确或不充分
脱节#5:未能将CAPA行动与设计控制联系起来
虽然这并不是一个全面的清单,但根据我们对最近FDA警告信的分析,我们认为这些是最重要的机会领域。我们建议您审查当前的质量管理体系,找出这些脱节之处,并确定补救措施,作为向QMSR过渡计划的一部分。
让我们回顾一下现行质量体系法规对设计控制的要求
FDA在21 CFR 820.30中规定了设计控制的现行要求。这些要求旨在涵盖医疗器械整个生命周期的设计和开发活动,并进一步分为10个子条款,如下图所示。

Figure 3: Summary of medical device design control requirements according to 21 CFR 820.30. Source: FDA.
值得注意的是,在适用于设计验证的21 CFR 820.30(g)的整个设计控制条款中,只有一项关于风险分析的要求:
Design validation shall include software validation and risk analysis, where appropriate.
事实上,在整个QS条例中,没有其他与风险有关的要求明确说明!不过,美国食品及药物管理局在QS条例的序言中提供了大量评注,以阐明其风险管理方法和对器械制造商的期望。例如,美国食品和药物管理局在序言中对第83条评论的答复中写道:
Risk analysis must be conducted for the majority of devices subject to design controls and is considered to be an essential requirement for medical devices under this regulation, as well as under ISO/CD 13485 and EN46001.
从整个医疗器械行业的普遍情况来看,风险管理的实践似乎没有达到美国食品和药物管理局的期望。如上所述,美国食品及药物管理局的检查意见经常指出许多不足之处,表明风险管理实践作为一门学科存在系统性差距。
部分原因可能是现行QS法规的编写方式,它将该局的执法权力限制在非常狭窄的范围内,即仅在设计控制下进行风险分析。另一个原因可能是,将风险分析与软件验证结合起来可能会给人一种印象,即这项要求只适用于软件,而不适用于所有设备。
正如ISO 13485和ISO 14971等当前最先进的国际标准所反映的那样,风险管理不仅仅是风险分析;它适用于质量管理体系的所有方面,而不仅仅是设计和开发。通过将ISO 13485: 2016作为规范性参考纳入QMSR,FDA将拥有在整个质量体系中执行基于风险的方法的监管权力。
ISO 13485:2016强调包括设计和开发在内的整个质量管理体系的风险
作为一项基本原则,ISO 13485:2016第4.1.2 (b)条要求采用基于风险的方法:
4.1.2组织应:
(b) apply a risk-based approach to the control of the appropriate processes needed for the quality management system.
显然,设计和开发是质量体系中的一个关键过程,其目的是提供安全有效的设备,同时符合适用的监管要求。因此,对设计和开发(第7.3条)提出了额外的基于风险的具体要求,这是ISO 13485 第7条所涵盖的产品实现过程的一部分:
The organization shall document one or more processes for risk management in product realization. Records of risk management activities shall be maintained (see 4.2.5).
此外,在第7.3.3条中,设计和开发投入:
Inputs relating to product requirements shall be determined and records maintained (see 4.2.5). These inputs shall include:
c) applicable output(s) of risk management;
在第7.3.9条中,对设计和开发变更的控制:
The review of design and development changes shall include evaluation of the effect of the changes on constituent parts and product in process or already delivered, inputs or outputs of risk management and product realization processes.
此外,应该理解的是,在需要进行规划(如设计和开发规划)的地方,隐含着一种基于风险的方法。
因此,ISO 13485要求将风险管理活动与设计和开发紧密结合起来。因此,按照现行QS法规的要求,仅关注风险分析已远远不够。这就要求我们转变观念,采用更周到、更系统的方法来培养风险管理方面的扎实能力。
现在,让我们从这个角度回顾一下风险管理与设计控制之间的5个潜在脱节之处
如上图1所示,设计验证是检查意见的首要类别,占FDA 2019年至2024年9月发布的1117条意见的30%。此外,被FDA定性为未进行风险分析或风险分析不足的风险分析缺陷占设计验证发现的45%。
以下是在最近的警告信中注意到的风险管理与设计活动之间的5个主要脱节之处。
脱节#1:风险评估中遗漏危害
在一封向心脏加热器-冷却器产品制造商发出的警告信中,FDA提出了以下意见:
Your firm has not adequately conducted risk analysis for your MCH devices, as required by 21 CFR 820.30(g). Specifically, your firm added a new hazard of “Bacteria such as m. chimaera or other biological agents being aerosolized into patient environments” to your MCH-10ARH Risk Assessment document on August 6, 2021; however, your firm has been aware of this hazard since at least 2018. Additionally, this hazard has not been considered as part of your design activities.
具有讽刺意味的是,FDA是在某些设计变更的背景下发出这封警告信的,这些设计变更的目的是通过增加一个可选的气流罩来降低感染风险。
Specifically, your firm has made significant changes that include, among other things, the addition of an optional airflow hood, a dripless external hose kit, and thermoelectric cooling technology in certain MCH models. The addition of an optional airflow hood is intended to reduce the risk of infection via aerosolization of contaminated water. The airflow hood impacts how potentially contaminated water droplets are dispersed in the operating room.
如果在风险评估中没有明确包括所有已知和潜在的危害,就有可能没有考虑足够的风险控制措施,也没有进行测试以确保这些措施有效。在本案例中,这种脱节也导致了对旨在控制细菌感染风险的变更设计验证不足。
要点:ISO 14971要求对风险分析、风险评估、风险控制措施的实施和验证以及总体残余风险的评估结果对每项已识别的危害进行追溯。
需要将风险分析(特别是危害分析)的结果与设计输入直接联系起来。将其视为危害、危险情况和危害的地图,以及它们如何与不同的设计输入相联系。行业最佳实践是开发一个有效的系统,将风险跟踪矩阵中的每个风险项目与设计输入跟踪矩阵中的一个或多个设计输入/输出相互参照。
脱节#2:并购后未更新风险分析
在向一家病人生命体征监护仪制造商发出的警告信中,FDA提出了以下意见:
Your firm has not adequately conducted a risk analysis for your nGenuity devices, as required by 21 CFR 820.30(g). Specifically, your firm provided a Hazard Analysis & Management document for your nGenuity CO2 Project, dated 2007, which was a document from the company you purchased in 2015. This did not identify risks associated with distorted displays or devices shutting down unexpectedly. Since 2016, your firm received 629 complaints related to distorted displays and at least 21 complaints related to monitors shutting down unexpectedly with your nGenuity device.
Also, the above 2007 Hazard Analysis & Management document for your nGenuity CO2 Project, did not identify any hazards related to faulty internal ECG cables, which were incorporated into certain nGenuity devices after 2015.
在同一封警告信中,FDA还列举了与设计验证和确认不足有关的其他意见。这些意见共同反映了对这些器械设计缺乏控制的严重关切。
关键点:并购(M&A)在医疗器械行业很常见。并购另一家公司后,器械制造商有责任继续保证被并购器械的安全性和有效性。这包括积极更新所有相关文件,尤其是与风险管理和设计控制相关的文件。
脱节#3:分析风险时对投诉数据分析不足
在向一家骨科植入物及配件制造商发出的警告信中,FDA提出了以下意见:
Failure to establish and maintain design validation procedures to ensure proper risk analysis is completed, as required by 21 CFR 820.30(g). Specifically, your firm’s procedure “Health Hazard Evaluation”, Document # 701-105-546, has not been adequately implemented to evaluate health risk(s). Your firm’s procedure describes requirements for calculating health risk and conducting an analysis of complaint data. The determination of criteria for the complaint search and analysis does not ensure that all applicable or potential failure mode codes are selected.
The complaint search criteria used in your HHE do not encompass failure modes identified in your Risk Assessment and Controls Report that would be conservatively included and analyzed in your firm’s HHE risk assessment.
在这种情况下,生产商仅使用了部分失效模式和危害来分析投诉,作为不符合材料规格要求或氧气透过率要求的聚乙烯肩部植入物包装袋的HHE风险的一部分。因此,他们遗漏了许多本应在分析中考虑的其他潜在失效模式。
关键点:ISO 14971第10条要求制造商建立一个系统,“actively collect and review information relevant to the medical device”,这些信息可能会影响安全性。投诉(包括不良事件报告)是必须分析的信息来源之一,最好采用有效的统计方法。有效的上市后安全监控系统对于信号检测、降低风险以及建立纠正和预防措施的反馈渠道非常重要。
脱节#4:对危害严重程度的定义不明确或不充分
在向一家领先的免疫测定分析仪和测定法制造商发出的警告信中,FDA提出了以下意见:
Failure to adequately establish and maintain procedures for risk analysis, as required by 21 CFR 820.30(g). Specifically, your firm's primary risk control procedure "Product Safety Risk Management," GLB-QS-PCD-0047, Revision 15.3, Dated 17Aug 2023, states that it is compliant with ISO 14971:2019 and EN 14971:2019 + A11:2021, and defines the(b)(4) severity of harm categories as: (b)(4). While the definitions for (b)(4) and (b)(4) as outlined in Appendix 9.1 are clear, the definitions of (b)(4), (b)(4), and (b)(4) are insufficient.
由于这封警告信中有许多相关细节被删节,因此很难理解本案的主要问题。因此,让我们来分析一下:
风险控制程序有5个不同的危害严重程度等级,这与当前的行业惯例一致。
其中两个严重程度等级的定义是明确的,但另外三个等级的定义不够详细,可能还包含相互矛盾的语言。
这种模棱两可的结果是,严重程度等级被错误地选择为用于对各种生命攸关的医疗状况(如乙肝感染、心脏病发作和治疗药物监测)做出治疗决策的检测的错误结果失败模式。
要点:ISO 14971将“风险”一词定义为发生危害的概率和危害的严重程度的组合。与ISO/TR 24971中的指导相一致,业界的普遍做法是对概率和严重性使用3级或5级标度。如果没有足够详细地定义这些等级,风险可能会因为分配不当而被低估或高估。正确估算风险对于评估风险可接受性以考虑风险控制措施(最好是通过设计)至关重要。业界的最佳做法是根据临床经验使用标准化术语和严重程度等级,以便进行更准确的风险分析。
脱节#5:未能将CAPA行动与设计控制联系起来
在向一家主要输液系统制造商发出的警告信中,FDA提出了以下意见:
Specifically, your risk analysis is inadequate in that it was not updated as required per Risk Management Procedure, 410-0009-01, Revision: 17.
The Ivenix Large Volume Pump’s Software Failure Analysis, 550-0015-04, Rev 2.0, did not include the hazard of extended start-up time to achieve the stated flow rate accuracy of +/-5%, nor was it revised to include this hazard after becoming aware of a product defect that allowed the device to exhibit this issue when infusing fluids at a rate of >50 ml/hr. and <200 ml/hr., as documented and identified in CAPA-00038 and FAI-4446. CAPA-00038 was initiated on April 1, 2022, due to startup time failures in high flow of the Ivenix Large Volume Infusion Pumps. Furthermore, this quality issue resulted in the issuance of a customer notification on April 22, 2022, and ultimately the Class II recall RES 92973.
在某种程度上,这种脱节与上文提到的脱节#1和#2类似。下面是另一个例子,即在发现新的产品故障时没有更新风险分析。这里的主要脱节是CAPA流程和风险管理流程之间的脱节,这似乎表明没有通过设计流程采取适当的纠正和/或预防措施。
制造商的应对措施是更新风险分析和对受影响人员进行培训。然而,食品和药物管理局认为这还不够:
Your responses are inadequate because they do not indicate that you plan to perform a retrospective review of CAPAs to ensure your risk analysis has been adequately updated, and there is no available evidence to document that your planned corrective actions will prevent recurrence of these violations.
要点:风险管理活动横跨质量管理体系的多个流程。必须了解这些流程的信息进出流程,以确保风险分析得到充分更新,从而推动采取适当的行动。这也是ISO 13485要求采用流程方法15进行质量管理的原因之一。不应将风险分析视为孤立、独立的活动,而应将其视为质量管理系统不可分割的组成部分。
风险管理活动与设计控制流程之间的脱节是医疗器械行业的一个重大问题。根据FDA的检查观察数据,45%的设计验证结果都源于风险分析。
在QMSR下,FDA对风险管理活动(尤其是在设计和开发方面)的审查只会越来越严格。这是因为对质量体系的关键流程采用基于风险的方法这一概念是ISO 13485: 2016的基本内容,该标准还概述了质量管理体系基于风险的具体要求。
通过研究最近的警告信,我们可以意识到风险管理活动与设计控制之间潜在的脱节。这些脱节通常会导致没有更新风险分析,而风险分析是设计流程的关键输入。如果风险管理流程的输出不能正确地在设计流程中流动,许多已知和潜在的风险就有可能得不到充分控制。这种脱节会对医疗设备的持续安全性和有效性造成严重后果。
认真审视风险管理过程,了解其输入和输出,以及它们如何与质量管理体系的其他过程(包括设计和开发过程)相联系,是非常有用的。

来源:MDR小能手


