您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-01-03 08:58
1、前言
鼻喷雾剂具有起效迅速、给药方便的优势,为生活中常用药物剂型,近三年鼻喷雾剂的销售额已超过100亿元。但目前国内申报获批的品种寥寥,原因究竟何在?鼻喷雾剂的药学研究存在哪些技术壁垒和挑战?CDE、FDA等监管机构对鼻喷雾剂的临床要求又如何呢?
2、鼻喷雾剂上市情况
目前申报仿制的鼻喷雾剂品种信息如表1所示,适应症基本集中于过敏性鼻炎。
表1 申报仿制的鼻喷雾剂品种情况
申报改良新药的鼻喷雾剂品种信息如表2所示。目前,广州新济药业申报的XJN010鼻喷雾剂已获批临床许可,是国内首款治疗帕金森鼻脑递送药物制剂。
表2 申报改良的鼻喷雾剂品种情况
目前鼻喷雾剂申报仿制和改良新药的品种数量仍然较少,预示鼻喷剂的药学研究和临床研究存在着一定的技术挑战。
3、药学研究内容
鼻喷雾剂一般分为溶液型和混悬型,在产品研发过程中需要结合产品特点对原辅料、包装材料(给药装置)进行充分的评估和研究。在仿制药开发的鼻喷雾剂应与参比制剂达成Q1/Q2/Q3一致。
关键原辅料质量控制 >>
鼻喷雾剂的原辅料研究需要根据剂型特点进行关键质量属性的控制,如混悬型鼻喷雾剂中使用的原料药,需要对粒度和粒度分布、颗粒形态、溶剂化物和水合物、晶型、溶解性等进行研究和控制。必要时需要将粒度和粒度分布和晶型列入质量标准;对于使用可影响混悬或颗粒性质等关键质量属性的辅料(如微晶纤维素-羧甲基纤维素钠等),需对质量标准进行额外的控制(如黏度等)。根据FDA相关指南,鼻喷雾剂关键原辅料建议研究至少3批次并与多批次参比制剂进行比对。
包材研究 >>
鼻喷雾剂包装材料一般由储药罐、喷雾泵、驱动器、 防尘盖等组成。包装材料的设计原理和使用方式与参比制剂保持一致,如喷雾泵剂量、驱动器喷孔的直径应与参比制剂相同,驱动器插入鼻腔部分的结构应与参比制剂相当。由于目前大部分原研制剂包材供应商受制于与原研厂家签订的排他协议,不能直接供货原研制剂包材,而包材的泵体结构的不同对喷雾性质影响极大,进而影响体外BE研究结果。另外提示一点,对于混悬型鼻喷雾剂,即使可以拿到原研的包材,由于自研药液与参比制剂药液的性质(如流变学相关性质)不完全一致,也很难达到体外BE等效的要求。因此鼻喷雾剂仿制药开发过程中需要针对自研制剂(一般采用中试批次样品)的性质,与包材厂家共同对包材进行结构调整,以达到体外BE等效的目的,同时,应考虑包装材料批间差异可能对产品质量产生的影响。新济药业前期在包材逆向剖析研究方面积累了丰富的经验,欢迎感兴趣的同行进行沟通交流。
质量标准研究 >>
溶液型鼻喷雾剂的质量标准研究项包括:性状、鉴别、pH 值、渗透压摩尔浓度、递送剂量均一性(瓶内、瓶间)、抑菌剂含量、有关物质、元素杂质、微生物限度、含量、装量及装量差异、每瓶总喷次等。混悬型鼻喷雾剂需增加对黏度、沉降体积比、原料药粒度分布等质量标准项研究。
体外BE研究 >>
FDA发布多款鼻喷雾剂的个药指南中明确需要进行体外BE研究,包括:每喷主药含量、喷雾雾滴粒度分布(雾滴分布)、小颗粒/雾滴中的药物量、喷雾模式、喷雾形态、启动和再启动(启喷和再启喷)。对于混悬液型的鼻喷雾剂,还需进行药物粒度分布和溶出度体外BE研究。
PBE研究项 >>
每喷主药含量
每喷主药含量测试应在产品使用生命周期的开始 (B) 和结束 (E) 阶段进行,每次测定的启动次数应为一次。
评价指标:自研制剂(3批)和参比制剂(3批)的每喷主药含量比较基于群体生物等效性 (PBE)分析。
喷雾雾滴粒度分布
使用激光衍射或经过适当验证的替代方法来确定喷雾液滴尺寸分布,仅需要在产品使用生命周期的开始 (B)和结束 (E) 阶段测量喷雾完全形成阶段(平台期)的喷雾液滴尺寸分布。建议选择距离驱动器喷孔 2~7cm之间的两个测试距离(间隔至少 3 cm)进行研究
评价指标:自研制剂(3批)和参比制剂(3批)在两个选定距离处对D50和SPAN(跨度)进行 PBE 分析。

小颗粒/雾滴中的药物量
建议在产品使用生命周期的开始 (B) 阶段,采用美国药典 (USP) <601> 规定的膨胀室装置或其他经过验证的高灵敏度测定方法测定小颗粒/雾滴中的药物。应使用最少的启动次数(通常不超过 10 次启动)来测定小颗粒/雾滴中的药物,以便更准确地反映单个剂量。
评价指标:自研制剂(3批)和参比制剂(3批)小于 9.0 µm 小颗粒/雾滴中的药物含量平均值进行PBE分析。

喷雾模式
喷雾形态是对喷射后特定延迟时间下(喷雾全部喷出后仍与驱动器喷孔接触时)的喷雾几何形状进行表征。喷雾形态应在产品使用生命周期的开始 (B) 阶段进行测试,可采用时间序列声控闪光摄影方法、激光成像技术、高速数码摄像机或其他适宜的方法进行测定。
评价指标:三批自研制剂与三批参比制剂(基于对数转换数据)的羽流角度和羽流宽度平均值之比应在90-111% 范围内。

启动和再启动
如果参比制剂说明书中有启动或再启动相关,则应在首次使用或其他条件(例如掉落)后存放指定的一段时间后进行再启动测试。
评价指标:在参比制剂说明书中指定次数的启动或重新启动之后,立即对单次启动每喷含量进行 PBE 分析。
药物粒度分布
对于双混悬型鼻喷雾剂,应使用显微拉曼设备来确定药物颗粒大小分布 (PSD)。
评价指标:自研制剂(3批)和参比制剂(3批)药物粒度分布的D50和SPAN(跨度)进行 PBE 分析。
体外释放
药物溶出是影响体内BE的最主要因素之一,采用具有区分力的体外释放方法,对产品使用生命周期的开始 (B) 阶段进行体外释放研究。
评价指标:使用相似性因子 (f2)、药物释放速率等参数进行判断。

稳定性研究 >>
鼻喷雾剂稳定性研究内容包括影响因素试验、加速试验和长期试验,必要时应进行中间条件试验考察,并应进行低温试验或冻融试验。如采用半渗透性容器,需设计低湿条件,并增加对失水率的考察。稳定性研究过程中,应考察在贮藏过程中易发生变化的,可能影响制剂质量、安全性和/或有效性的项目。若处方中含有抑菌剂、稳定剂等辅料,在稳定性研究中还需考察上述辅料含量的变化情况。
4、临床研究内容
根据鼻喷雾剂自身性质的差异,临床研究的内容有所差异。参考FDA指导个案和CDE指导原则:对于溶液型的鼻喷雾剂,一般可通过Q1/Q2/Q3一致的方式豁免BE;对于混悬型的鼻喷雾剂,一般需要通过“Q1/Q2/Q3一致+PK-BE+临床终点-BE”的方式达成BE。FDA对鼻喷雾剂的指导个案以及部分已上市鼻喷雾剂品种在国内开展临床试验的情况如下(表3、表4)。
表3 部分鼻喷雾剂产品的FDA指导个案推荐BE方案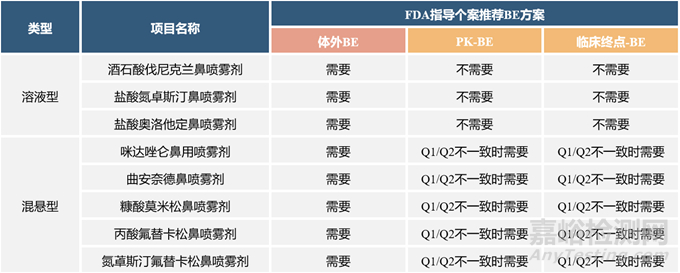
表4 国内目前已获批的鼻喷雾剂的临床试验登记情况
参考文献:
1.国家药监局药审中心,国家药监局药审中心关于发布《化学药品吸入液体制剂药学研究技术要求》的通告(2021年第47号),2021.11.25,https://www.cde.org.cn/
2.国家药监局药审中心,国家药监局药审中心关于发布《化学药品仿制药混悬型鼻用喷雾剂药学研究技术指导原则》的通告(2024年第28号),2024.06.07,https://www.cde.org.cn/.
3.FDA. Guidance for Industry-Bioavailability and Bioequivalence Studies for Nasal Aerosols and Nasal Sprays for Local Action[EB/OL]. [2023-06-26]. https://www.fda.gov/ media/70867/download.
4.FDA.Product-Specific Guidances for Generic Drug Development[EB/OL]. [2023-06-26]. https://www.accessdata. fda.gov/scripts/cder/psg/index.cfm.

来源:Internet


