您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-02-14 20:20
溶解度是指可以溶解在指定体积溶剂中物质的最大质量。以定量术语而言,溶解度被定义为一定温度下饱和溶液中溶质的浓度。当溶质与固相(溶质)处于平衡状态时,认为溶液是饱和的。溶解度是影响药物吸收和全身循环水平的重要因素之一。药物的 BCS 溶解度类别是通过将单一剂量单位药物溶解于 250 mL、pH 1.2~6.8 的缓冲液中进行测定的。250 mL 的测试体积是根据典型的生物等效性研究方案得出的,该方案建议给空腹的志愿者服用八盎司(240 mL)水,并考虑 10 mL 的胃静息体积。
当溶液的剂量 / 可溶体积≤250 mL,即剂量分数(D0)≤1 时,认为该药物是高度可溶的。水溶性差是药物发现与开发中的主要挑战之一,因为药物必须在吸收部位溶解才能被吸收。在不同的药典中,使用不同的描述性术语来表示溶解度的范围类别(表 1)。这种传统方法的问题之一是忽略了剂量,因此一个高活性的化合物根据其理化特性可以归类为不溶,但是由于其剂量很低,仍可能完全溶解在相应体积的溶剂中。相反,BCS 考虑了所需的剂量,通过使用 D0 进行溶解度分类,避免了理论定义与实际行为之间的不匹配性。
表 1 药物溶解度级别划分
|
|
|
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1、 溶解度和溶出度
溶出是指固相(片剂 / 粉末)进入溶液相(如水)的过程。本质上,当药物溶出时,固体颗粒会分离并逐个分子与液体混合并成为液体的一部分。将片剂引入溶液中时会发生药物的溶出,并通常伴有固体基质的崩解和解聚,随后药物从剩余的小颗粒中扩散。口服水溶性较差的药物时,药物在胃肠道中的溶解度差和溶出慢通常会导致吸收和生物利用不足 。普遍采用诺伊斯 - 惠特尼(Noyes - Whitney)方程来表达不同因素对于溶出的影响 :
其中 A 为固体药物的表面积,D 为药物的扩散系数,h 为与溶解表面相邻的有效扩散边界层的厚度,Cs 为药物的饱和溶解度,V 为可用的水量,Xd 为溶解的药物量。
随着颗粒粒径变小,A 随着粒径的减小而增加,引起溶出速率(dXd /dt)变快。改变溶剂的 pH 值也可能会对可离子化药物(碱性和酸性)的饱和溶解度(Cs)产生影响,可能导致其溶出速率增加或降低。此外,溶出度很大程度上受药物的理化特性和胃肠道各个生理因素的影响。
对于 BCSⅡ 类或 Ⅳ 类药物,肠道吸收可被视为受限于溶解度和溶出度。Ⅱ 类药物是低溶解度、高渗透性的化合物,可定义为具有高吸收分数(An)和大于 1 的剂量分数(D0)。当此类化合物的溶出度偏低时,溶出分数(Dn)小于 1,而 An 和 D0 较高。另一方面,如果 An 和 Dn 都较低,则可将该药物划分为 BCSⅣ 类。
当肠道吸收受到溶出度或溶解度限制时,胃肠道中药物的浓度将由相关限制因子来调节。由地高辛(digoxin)和灰黄霉素(griseofulvin)的经典案例可知,溶出度和剂量影响高渗透性药物的吸收剂量分数(fraction of dose absorbed,Fabs)。地高辛和灰黄霉素的溶解度非常相似(约 20 mg/mL),但其剂量却大不相同(地高辛为 0.5 mg,灰黄霉素为 500 mg),地高辛具有较低的剂量分数(0.08),而灰黄霉素的剂量分数较高(133)。因此,溶解 1 单位灰黄霉素需要超过 33 L 的水。仅仅是胃肠道内的液体不足以溶解这一剂量,所以灰黄霉素显示出高剂量分数和低溶出分数。可以通过降低给药剂量、服用更多液体,或增加药物溶解度来增加药物吸收剂量分数和生物利用度。
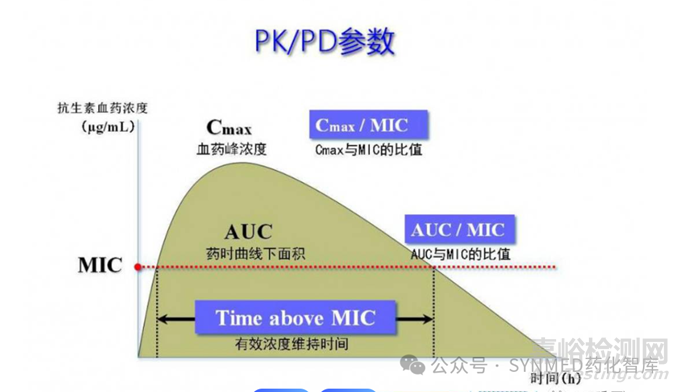
由于药代动力学(pharmacokinetic,PK)和药效学(pharmacodynamic,PD)因素,灰黄霉素的给药剂量不能改变,而溶解药物所需水的量又受到胃部结构和生理功能限制,因此只能通过合适的制剂改善其溶解度,这也是唯一可减少 Dn 并增加口服吸收的策略。另一方面,就地高辛而言,问题在于其动力学性质,从低 D0 可以看出该药物可以完全溶解,但由于粒径大小方面的限制,药物的溶出可能会非常缓慢(Dn<1),这也限制了药物在胃肠道环境中的溶出量和总吸收量。据计算,大于 10 μm 的粒径将导致吸收受到溶解速率的限制。因此,如果减小粒径,则可能实现肠道的完全吸收。实际上,微粉化的地高辛粉末具有足够的溶解速率,并且较长的肠道停留时间使其足以完全吸收。相反,灰黄霉素的吸收受到溶解度限制,并且改善其溶出度不能显著增加吸收剂量分数,微粉化也不能显著增加灰黄霉素的吸收。因此,需要开发可在胃肠道环境中充分溶解的可溶性制剂。
2、 Log P
决定药物水溶性的主要因素之一是药物与水分子形成氢键的能力 。高水溶性对于药物在水性介质中的溶解是有利的,但与此同时,由于具有高极性和亲水性,这些化合物通常显示出低渗透性。脂水分配系数(lipo - hydro partition coefficient,Log P)是表征化合物在亲脂性(正辛醇)和亲水性(水)溶剂中溶解度的参数,具体为两者比值的对数,基于此可以根据药物的亲水性或疏水性对其进行排名。除了以正辛醇 / 水分配方法来衡量亲脂性外,也可以通过化合物的动态能量性质来描述。
3、 pH
由于溶剂 pH 对药物离子化的影响,药物从脂溶性环境向水溶性环境的分配能力可以定义为与溶剂 pH 相关的函数。通常,离子化药物具有比非离子化药物更好的水溶性。因此,可溶性离子在水性介质中的溶解速率可能会受到溶剂 pH 变化的影响。亨德森 - 哈塞尔巴尔赫(Henderson - Hasselbalch)方程用于描述 pH 对药物电离的影响:
弱酸:未解离(%)= 100 / [1 + anti - lg(pH - pKa)]
弱碱:未解离(%)= 100 / [1 + anti - lg(pKa - pH)]
弱碱性化合物在高于其解离常数的 pH 值下可能具有较慢的溶解速率,因此更多的药物分子以结合的形式存在。另一方面,弱酸性化合物在高于其酸解离常数的 pH 值下会显示出更快的溶解速率,更多的药物分子呈离子化形式。胃酸的生理 pH 值为 1.4~2.1,其受食物摄入量的影响很大,范围从 1~8 不等。
小肠的 pH 值高于胃,食物的存在对其影响不大。小肠 pH 值显示从近端(十二指肠)到远端(回肠)段的梯度上升。肠道的 pH 值范围为 4~8 。食物摄入后胃 pH 值的升高会增加碱性药物非离子化形式存在的比例,并降低药物的溶解速率。例如,如果在进餐过程中提高胃液的 pH 值,则可使弱碱性药物茚地那韦(indinavir)(pKa 值分别为 3.7 和 5.9)发生沉淀,与禁食的受试者相比,会导致其 AUC 和 Cmax 值显著降低 。另一方面,食物也可以通过增加药物的离子化程度来提高弱酸性药物的溶出度,如布洛芬(ibuprofen)。
4、 胆汁酸盐
胆汁酸盐(bile salts)是胆固醇的衍生物,为两亲性甾体类生物表面活性剂,在肝脏中生成并储存在胆囊中。胆汁酸类固醇骨架的凹面由于存在羟基而具有亲水性。然而,由于角甲基的存在,其凸面是疏水的。这种特殊的结构使其不同于传统的表面活性剂,后者通常含有极性头部和较长的非极性链。胆汁酸盐的润湿作用和胶束化作用可能会显著影响低溶解度药物的溶解度和溶出度。超过临界胶束浓度(critical micelle concentration,CMC)时,胆汁酸盐会聚集并形成胶束,通过形成亚微米级混合胶束,增加了亲脂性药物的溶解度,使疏水性分子更易溶解,进而更可能到达肠黏膜上皮细胞。例如,当胆汁酸盐的浓度随胆汁酸浓度的增加而增加时,亲脂性药物利福昔明(rifaximin)的溶解度也随之增加,从而增强了其抗菌作用。
5、 粒径
化合物的粒径是影响溶解速率的重要物理参数。根据诺伊斯 - 惠特尼模型,粒径越小,表面积越大,进而溶解速率也更高。颗粒的密度也会影响溶出度,因为密度会改变体内颗粒的分散度,而更好的分散也会增加溶出度。据报道,在禁食条件下,粒径较大会极大地影响可溶性差的抗逆转录病毒药物的溶出度和口服药物的吸收,而在餐后(进食)状态则无此作用。自 20 世纪 80 年代以来,纳米化(粒径减小至纳米级)引起了相当大的关注,特别是其可增加亲脂性药物的生物利用度。许多研究已证实了粒径与溶解度、溶出度和生物利用度的相关性。
为了改善药物在肠道的吸收,现代生物制药方法使用非晶态固体分散体(amorphous solid dispersion,ASD)技术,可以使药物在适当的时间段内达到并保持过饱和状态。通过使用不同类型的聚合物,可以抑制难溶性药物形成晶体,从而保持过饱和状态并有助于避免析出。聚合物具有调节粒径的能力,能够重新溶解并降低所析出活性药物的粒径,从而达到更好的肠道吸收并改善其生物利用度。
对于药物粉末,溶出介质的可用表面积比粒径更为重要。当高疏水性药物在溶出介质中的润湿性能较差,并且由于制备工艺改变粒径而改变溶出度时,这一点尤为重要 。
有关粒径减小策略及其对吸收影响的详细讨论,可以另起一篇写,这里暂时不作讨论。
6、 液体体积
胃肠道中的液体体积取决于与药物合用的水量、胃肠道内的分泌物量,以及整个肠壁的液体通量。在体外,采用生理学上适用的溶解液研究药物的溶出速率和水平,可帮助研究人员更好地理解和预测药物的体内吸收。研究表明,较小的胃液量可能会降低硝苯地平(nifedipine)的溶出,并减少其在人体内的吸收。采用 GastroPlus 进行的高级计算分析表明,对于水溶性较差的药物,肠液体积对其平均血药浓度曲线的预测具有很大影响。
慕迪(Mudie)等量化了胃和小肠的总体积和水分布,并揭示了小肠中存在不连续的液体囊 。这项研究表明,小肠远端区域中液体的百分比最高(远端十二指肠、近端回肠和远端回肠),因此十二指肠和空肠近端是吸收的主要部位。当然,药物需要先溶解,然后才能到达小肠黏膜。

来源:SYNMED药化智库


