您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-03-09 17:50
摘 要 Abstract
美国食品药品监督管理局(FDA)对药品、医疗器械的监管重点逐渐从传统上重视上市前审批向上市后监管转移,覆盖产品全生命周期,并显示出逐渐从以还原论为中心向整体论优势转型的趋势。FDA 在美国联邦机构中率先尝试应用包括深度学习(DL)在内的人工智能(AI)技术。在批准越来越多采用AI 技术产品的同时,FDA 还尝试将AI 用于监管机构内部工作,包括从外部招聘专业人才、面向行业设立开放式挑战项目与试点计划,强化监管机构内部AI 能力建设与专业知识水平提高。AI 的快速发展加快了FDA 监管范式转型。与AI 发展类似,医药产品监管同样面临不确定性。本文基于FDA在相关领域的具体实践,总结了AI 助力FDA 监管范式转型的作用与发展趋势,以期为其他监管机构在相关领域的发展及相关技术的应用提供有益的经验。
The U.S. Food and Drug Administration’s (FDA) regulatory focus on pharmaceuticals and medical devices has gradually shifted from the traditional emphasis on pre-market approval to post-market supervision covering the entire product lifecycle. This shift shows a trend from a reductionist approach to the advantages of a holistic methodology. The FDA was the first federal agency to experiment with artificial intelligence (AI), including deep learning (DL). In recent years, while approving a growing number of products that incorporate AI, the FDA has also experimented with using AI for internal purposes—such as recruiting external experts, launching open challenge initiatives and pilot programs for the industry. The rapid development of AI has accelerated the FDA's regulatory paradigm transformation. Furthermore, the regulation of pharmaceuticals and medical devices also faces uncertainties, and the FDA's exploration in these areas can offer valuable insights for other government agencies seeking to apply similar technologies. This paper summarizes the role and development trends of AI-assisted regulatory paradigm transformation at the FDA, based on its specific practices, to provide references for other regulatory agencies in related fields.
关键词 Key words
人工智能;范式转型;锁定算法;内部能力建设;不确定性;智慧数据
artificial intelligence; paradigm transformation; locked algorithms; internal capacity development; uncertainties;smart data
2017 年, 时任美国食品药品监督管理局(FDA)数字医疗部门负责人Bakul Patel 公开招募13 名工程师,其中包括软件开发人员、人工智能(artificial intelligence,AI)与云计算专家等。Patel 希望借助依据《医疗器械行业付费修正案》(Medical Device User Fee Amendments,MDUFA) 成立的数字医疗卓越中心,重组FDA 对数字医疗的监管模式, 使得新的解决方案和技术更容易、更快进入市场。其中一个设想是让审批过程更类似于美国运输安全管理局(Transportation Security Administration,TSA)的安检线,即值得信赖的公司接受更为宽松的监管,而存在历史不良记录的公司将接受更彻底的审查。Patel 希望,相关联邦法规不会阻碍与FDA 过去监管的医疗器械截然不同的创新产品的发展[1]。尽管2022 年Patel 已从FDA 离职[2],但在FDA 内部,AI 正在彻底改变药品开发、医疗诊断和医疗服务等的模式[1]。
在美国行政会议(Administrative Conference of United States,ACUS) 委托撰写的题为《算法政府:联邦行政机构中的AI》(Government by Algorithm: Artificial Intelligence in Federal Administrative Agencies)报告中,展示了FDA 如何在多个政府机构中率先尝试使用包括深度学习(deep learning,DL)在内的先进AI 技术[3]。该报告预测,FDA 采用人工智能/ 机器学习(artificial intelligence/machine learning,AI/ML) 工具将带来从上市前审批向上市后监督工作的更广泛转变。
1. FDA 处于公众健康和安全领域AI 快速发展的前沿地位
FDA 正处于公众健康和安全领域AI 快速发展的前沿地位,处于监管范式转型之中,AI 的飞速发展进一步推动了这一转变。但由于近年来医药产品的安全性和有效性标准受到诟病, 公众对FDA 的信任度下降,以及AI 技术对FDA 透明度、问责制、客观性和合法性造成的威胁等,AI 推动的监管范式转型面临诸多挑战。FDA 通过在AI 监管能力方面持续投入,利用AI 加快和扩展药品和医疗器械的使用,同时保持医药产品安全的“金标准”,逐步提高公众健康和安全领域的变革潜力。
FDA 在美国联邦机构中率先尝试应用包括DL在内的AI工具[4]。具体用例包括基于AI 的重复数据删除算法对重复报告进行分类,该算法适用于FDA 不良事件报告系统(FDA Adverse Event Reporting System,FAERS)中的非公开数据, 用于识别确定重复报告。首先, 采用自然语言处理(natural language processing,NLP) 系统处理FAERS 自由文本叙述中的非结构化数据, 提取相关的临床特征;然后,将结构化和非结构化数据用于概率式记录关联方法(probabilistic record linkage),通过评估多个数据字段(datafield)和应用每个字段的相对权重,对报告进行评分。最后,可能重复报告的输出结果将被进一步分组,以便在病例系列评估过程中识别和确认FAERS 报告,并认定有必要加以关注的安全问题。
2. AI 的快速发展正在影响FDA 监管
AI 的快速发展正在以两种方式影响FDA 监管:一方面,越来越多的创新产品融合了AI 技术,给FDA 带来了更多的监管挑战;另一方面,FDA 尝试将AI 用于内部工作。
2.1 AI 医疗设备监管
AI 在医疗领域的应用为医疗实践带来了革命性变化。AI 模式识别使AI 医疗设备能够识别和确认某些类型的伤害,有助于临床诊断病症,改善医疗成像质量,并预测将来可能发生的不良医疗事件[5-7]。
在Viz.AI Contact( 用于检测卒中)、OsteoDetect(用于识别骨折)和IDx-DR(用于识别糖尿病视网膜病变)的制造商证明了具体的性能标准之后,FDA通过针对中风险、低风险新型设备的替代审批通道——De Novo审评流程,批准了这些设备上市。针对这些首次获得批准的AI 医疗设备,FDA 有必要寻求临床决策支持产品相关的监管框架和方案,进而鼓励开发者创建、调整和扩展软件的功能,辅助医疗机构诊断和治疗疾病。
下一代AI 医疗设备将带来更多的监管挑战。迄今为止,获得FDA 批准的相关设备均为“锁定(算法的)”设备,这些设备依赖于制造商更新,而非独立地根据观察到的新数据做出调整。FDA认识到,除了在医疗保健领域应用成熟的锁定算法外,还应有很多预期,使AI 模型能够随着时间推移而进行动态更新,有望解锁更好的性能和更个体化的医疗保健服务[8-9]。2019 年,FDA率先在讨论文件中提出“预定变更受控计划”(predetermined change control plans,PCCP)的概念[10]。2021 年1 月,FDA发布《作为医疗器械的软件(SaMD) 行动计划》[Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) Action Plan, 以下简称SaMD 计划],概述了PCCP流程,制造商可以通过该流程申请某些类型AI 医疗设备动态更新的预先批准[11]。预期FDA 将会尝试批准解锁算法的动态AI 设备。关键在于,FDA 在SaMD计划中强调,需要在上市前开发到上市后的全过程监控所有动态AI 医疗设备性能[11]。
医疗器械行业游说组织美国先进医疗技术协会(Advanced Medical Technology Association,AdvaMed)建议,传统上在临床环境中使用的510(k) 医疗器械,如果变更用于居家使用,应在专门的510(k) 通道下审查。此外,AdvaMed 还要求FDA 使用国会授予的PCCP 权限。AdvaMed表示,对于大多数经FDA 批准在临床环境中使用的医疗器械,如果变更使用环境,就必须提出新的上市申请,这是一个耗时且成本高昂的过程。为促进医疗器械及时扩展到居家和其他非临床环境使用,AdvaMed 建议, 对那些已经获得FDA 批准在其他环境中使用的医疗器械,FDA 可采取更高效、简化的审查方法[12]。2024 年8 月,FDA 发布关于医疗器械PCCP 的指南草案,详细说明了FDA 认为医疗器械的哪些变更适合PCCP,以及申办方应在其上市前批准(premarket approval,PMA)、重新批准或510(k)申请中提交的具体资料清单[13]。2024 年12 月,FDA发布PCCP 指南定稿, 详细说明针对AI 医疗设备使用PCCP的预期[14]。FDA 重申,ML 是AI 的一个子集。指南草案侧重于针对ML 开发提供一种前瞻性思维方法,而指南定稿则采取了一种更广泛的方法,并表示其目的是为更广泛的AI 设备软件功能(artificial intelligence-enabled device software functions,AIDSF)服务[14]。依据PCCP,制造商可以通过采集数据、重新训练AI 模型并开展测试,来证明误报率已显著降低;同时需更新设备的标签说明,并向用户传达变更内容。由于相关变更依据PCCP 进行,制造商无需提交新的上市申请。然而,如果对AI 模型进行修改,使其能够在生理不稳定(physiological instability)发生之前提前预测,而此前的版本无法做到这一点, 则不属于PCCP 的范围,针对相关变更需提交新的上市申请。
2.2 FDA 内部AI 应用与维护
FDA 科学家在科学研究和监管决策中经常使用建模和模拟的方法[15]。此外,FDA 还使用虚拟家庭和特定患者模型以及模拟临床试验,以降低志愿者参与临床试验的风险与成本[16-19]。除了针对受监管产品开发与具体产品的相关用例外,FDA 利用AI 在日益复杂和高风险的监管环境中更有效运作的关键在于,FDA 需要将AI 付诸应用,通过在日常监管中采用AI 实现获益。
FDA 采用AI 模型挖掘数据,用于认定药品与相关病症之间的新关系,协助药品安全相关的上市后监管。FAERS 数据库包含提交给FDA 的不良事件报告、用药差错报告和导致不良事件的产品质量投诉[20-22],这些报告和投诉均为自由文本格式[23]。
FDA 主要通过510(k) 医疗器械许可流程监管作为医疗器械的软件。在管理和临床环境中使用的AI 应用程序必须遵守美国国家卫生信息技术协调员办公室(Office of the National Coordinator for Health Information Technology ,ONC)的规定,例如强制要求算法透明[23]。值得注意的是,直接面向消费者的涉及医疗保健的AI工具,属于消费品的管辖范围,这一新兴领域几乎没有强制监管。FDA 的医疗器械监管始于1976年,主要目的在于监管硬件设备,而不是针对依赖于训练数据且需要精细、持续性监控的软件。同时,1996 年发布的《医疗保险可携性与责任法案》(Health Insurance Portability and Accountability Act,HIPAA)在数字医疗信息爆炸式增长之前制定,当时难以预见到需要大量电子病历来训练ML 算法[24]。因此,相关人士认为,现行的监管框架已经过时。医疗保健领域的传统监管范式迫切需要适应AI 快速发展。要想实现医疗保健领域AI 的有效治理,必须建立新的监管框架或大幅改变现行的监管框架[25]。
FDA 与斯坦福大学的数据科学家合作测试了不同的NLP 模型, 以预测FAERS 报告包含药品安全问题政策相关信息的概率[26-27]。根据世界卫生组织- 乌普萨拉监测中心(World Health Organization-Uppsala Monitoring Centre,WHO-UMC)的药物报告训练模型,由FDA 安全评价员人工标注报告,用于药物因果关系评估。FDA 认为,该试点项目可作为系统改进的依据,有利于优化监管资源配置,识别并确定上市后安全问题[4]。在第二个试点项目中,FDA 使用类似的NLP 技术将FAERS 的非结构化数据转化为结构化数据,然后尝试建立不同药品与肝功能衰竭之间关系的模型。与FDA 和斯坦福大学合作试点项目相比,第二个试点项目在确定FAERS 审查的优先次序方面没有达到预期,未能证实或反驳FAERS 报告中涉及药品与肝功能衰竭存在因果关系,也没有产生足够准确的输出结果。在这两个试点项目中,FDA 比较了多个NLP 模型的性能,包括神经网络、逻辑回归、随机森林和支持向量机模型,优化了模型参数,使得在某些情况下预测准确率超过90%。
FDA 充分认识到,相关领域的AI 模型需要长期迭代、反复实验。2020 年初,precisionFDA启动“ 使用FDA 开放数据检测不良事件异常值获得新见解”(Gaining New Insights by Detecting Adverse Event Anomalies Using FDA Open Data) 挑战, 要求参与者开发AI/ML 模型,以便更好地从FDA的FAERS 报告中检测可能存在的安全问题[28]。与湿性实验室科学挑战相比,设立基于计算的挑战的启动成本较低,因此这类相关的AI 模型创新更适合公众参与。precisionFDA 挑战中AI 模型创新相关项目[28-33] 见表1。

除了针对NLP 技术的投入外,FDA 还启动重组数据业务和重塑其技术数据基础设施的计划, 例如信息交换和数据转换(INFORMED)计划,该计划责成具有医学专业知识的常驻企业家、工程师和数据科学家制定战略,确定FDA 应如何在大数据分析能力方面投入[34-35]。该计划的目标包括继续扩展和维护数据科学和大数据分析的组织和技术基础设施;支持肿瘤药物监管科学研究中的系统思维,制定新的解决方案,提高相关工作流的效率、可靠性和产出效率。
生物医药领域内的大数据在数据量(数据大小)、变异性(数据类型)、真实性(数据噪声和不确定性)和速度(数据流和处理)4 个维度的特点见图1。目前,FDA 的审评审批决策通常基于种类有限的数据,主要来自临床试验和临床前研究;这些数据大多是结构化的;数据集的大小通常不超过几千兆字节;作为注册申请的一部分,不具备连续性。生物医药领域内的大数据在4 个维度的扩展要求持续提高技术能力和组织能力。预期未来将通过综合考虑患者、疾病和具体的环境特征,将大数据转化为智能数据,实现个体化治疗。
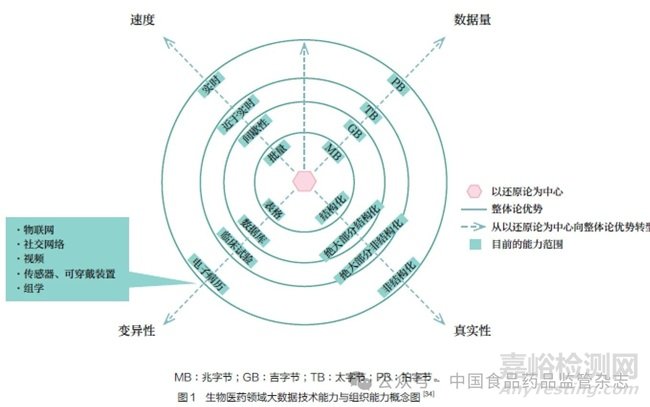
3. FDA 监管范式转型:从上市前审批到上市后监管
FDA 用于挖掘大数据的AI技术创新可能会推动监管范式转型,即从投入大量资源和精力专注于严格的上市前审批,转向更动态、更严格的上市后监管。这种转变不仅在促进创新方面大有可为,还在AI 支持的上市后监管机制中有望实现更有效的机构合作和监管执法。其主要优势体现在:① AI 技术有利于提高FDA决定对企业采取调查和整改行动的速度。②有利于引导企业建立健全算法监控系统,提高产品和制造质量,进而使公众受益。
3.1 FDA 传统监管范式
FDA 传统上属于严格的事前监管机构。FDA 的传统监管框架对药品和医疗器械实行严格的上市前审批,而上市后监管相对有限。
以新药为例,上市前审批程序的核心是新药申请程序,要求申办方开展3 期上市前临床试验,证明相关药品的安全性、有效性符合FDA 的要求。在传统监管范式中,作为确保药品、医疗器械安全、有效、质量可控的核心部门,FDA 一直非常重视最大限度减少“第一类”误差(假阳性)或批准最终发现存在安全问题的药品,而以增加“第二类”(假阴性)误差为代价,推迟或阻止实际上能为患者带来净获益的药品上市。部分人士认为,这样的传统监管范式使FDA 积累了巨大的声誉资本。但随着加快审批受到越来越多的关注,外界对近年来FDA 的声誉资本受到侵蚀深表关切。
尽管FDA 要求分3 期开展临床试验,但传统的临床试验模式只能在相对较短的时间内提供相对有限的人群信息,最有可能发生不良反应的人群通常被排除在临床试验之外。了解获益与风险是一项持续过程,随着上市后数据的积累,更新获益- 风险评价面临巨大挑战(图2)。在对利用AI 技术模拟临床试验改善安全结果的可能性持开放态度的同时,应该认识到临床试验有可能掩盖的严重的安全风险。

传统临床试验通常在与临床或居家环境不同的特定人群和专业环境中开展。这些试验可能会采取一系列旨在控制变异性并确保其产生的数据质量的措施,例如制定入组标准,使用与普通病历分列的详细病例报告表,详细定义研究流程,以及监测专业研究人员,确保其遵守研究方案,确保数据采集的准确性等。
现阶段随机对照试验仍然是建立医药产品安全性、有效性科学证据的有力工具,同时能让研究者和监管机构了解相关产品的治疗作用所涉及生物学机制。临床试验可为医药产品的上市前评估提供证据以证明相关疗法可能有效,但其存在以下局限:①传统临床试验有严格的入组标准,参与临床研究的人群可能与真实世界中使用医药产品的人群存在差异,因此通过这些临床试验获得有效性数据,通常以更多受众在实际使用中的不确定性为代价。②传统临床试验的数据采集往往出于满足研究需要的目的,可能很少有伴随疾病和治疗相互作用的数据。③基于传统临床试验获得的证据可能在及时性与相关性方面存在欠缺,不足以弥补很多证据缺陷,可能降低相关证据在真实世界决策中的可推广性。④多年来,开展大型传统临床试验的费用一直在稳步增长。近期估算表明,传统临床试验的实际成本轨迹可能会急剧增长,但支持医疗照护决策的证据数量并没有相应增加[36-37],如图3 所示。⑤与产品开发阶段和上市后的随机对照试验相比,上市后监测与观察性研究采集的数据量更多[38],如图4 所示。
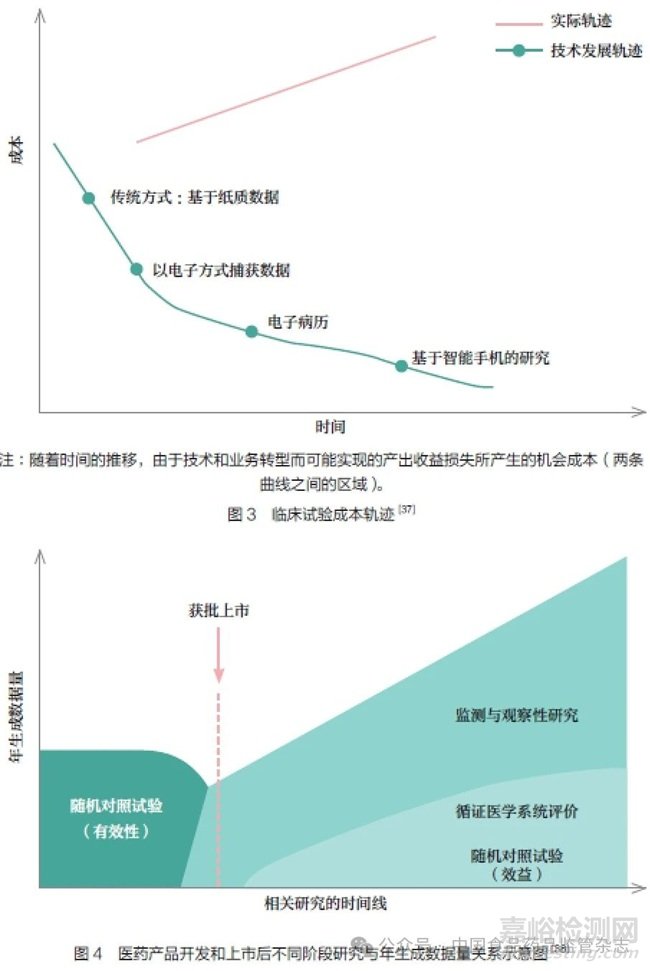
由于随机对照试验的样本量相对较小,以及在招募研究参与者时存在选择偏倚,对于少数族裔和边缘化社区人群,风险尤其高。如果不加以注意, 使用AI可能会加剧这类偏倚。模拟试验可以作为随机对照试验的补充(而非替代),允许重复开展试验,或降低非代表性临床试验中固有的种族差异风险。开展模拟试验的科学家可以主动在与其模型互动的虚拟队列中建立代表性。此外,通过新药创新科技方法(Innovative Science and Technology Approaches for New Drugs,ISTAND)试点计划,以及作为FDA 模型引导的药物开发配对会议计划(Model-Informed Drug Development Paired Meeting Program,MIDD)的一部分,利用基于AI的模型弥合某些未研究的患者亚群或使用场景的有效性和安全性后,有望实现更有效地采集临床试验数据,同时更好地代表不同的患者群体。
除了FDA 现行上市前审批成本高昂和可能不具代表性外,这种严格的事前监管方法还可能会延长创新药品和医疗器械产品的上市时间[39]。迄今为止,FDA 仍在“第一类”和“第二类”误差之间权衡,不可能在减少一种误差的同时不增加另一种误差。即使是态度最为坚定的批评人士也认识到,如果FDA 将关注的重点从严格的上市前标准转移至上市后监管,就需相应增加用于上市后监管的资源[40]。近年来,FDA一直在逐步完善上市后监管制度,而AI 领域的快速发展有望带来更具变革性的转变。
3.2 AI 加速FDA 向上市后监管转型
AI 技术的快速发展极大推动了FDA 的监管范式向上市后监管转型。在过去几年里,FDA 对上市前测试的严格要求有所放宽,实际上逐步将审查资源从上市前向上市后倾斜[41-42]。
2007 年颁布的《食品药品管理法修正案》(Food and Drug Administration Amendments Act,FDAAA)授权FDA 监测已获批药品的安全风险,同时要求制药企业开展上市后安全研究[43]。近年来,为保证加速审评通道的正常发展,多位国会议员要求FDA改革加速审评通道,增加上市后监测资源配置。
FDA 持续探索使用AI 工具提高大规模评价上市后数据的能力。自2000 年以来,FDA 已逐步将来自制药企业的大量数据全部汇集到不良事件报告数据库(包括FAERS)中,为上市后监管提供信息。在极少数情况下,FDA 甚至会根据对上市后监测数据的深入了解来重新评价上市前的审批决定[44]。
在将AI 应用于上市后监测制度以后,2018 年,时任FDA 局长Scott Gottlieb 宣布在处理积压的上市后监测分析工作方面取得了显著进展[45]。FDA 在其历史上的大部分时间里都专注于上市前审查,现阶段已逐渐将上市后监测作为一个越来越重要的优先事项,并且认识到这种持续的审查是FDA 职责的关键部分[46-48]。尽管使用NLP 模型分析FAERS 数据成为FDA 迄今为止在上市后监测方面的主要尝试,但FDA 更加关注上市后监测的监管范式转型,未来可能扩展到更多的数据源,并利用包括大型语言模型在内的其他AI 技术[49-52]。
3.3 对监管范式转型的初步评估
随着新一代包含动态AI 的医疗设备的出现,其带来的监管挑战使得FDA 监管范式向上市后监管的转变成为实际需要。FDA关于新型AI 医疗设备的监管指南强调了AI 模型上市后监测的必要性。2021 年10 月,FDA、加拿大卫生部(Health Canada)和英国药品和健康产品管理局(Medicines and Healthcare Products Regulatory Agency,MHRA)共同发布《医疗器械开发机器学习规范》(Good Machine Learning Practice for Medical Device Development: Guiding Principles),上市后监测成为其关键原则之一[53]。
FDA 加强上市后监管为实现更精简、更宽松的上市前安全审查提供了可能,有利于保持甚至提高整体安全性。例如,在应用AI 软件的医疗器械监管领域,FDA 正在尝试制定更为精简的上市前审查方案,同时持续加强上市后监管。FDA 放宽上市前临床试验研究的事前要求,允许AI 驱动的模拟模型,虽然可能会导致药品审批中的“错误结论”(即前文所述“第一类”误差)[54-55],但在FDA 同时加强上市后监管的背景下,预期能够缓解相关风险。
监管机构应该考虑放宽上市前监管要求能在多大程度上降低“第二类”误差,在促进创新的同时尽量缩短上市周期,避免因为增加上市后监管资源配置而牺牲整体安全性(即“第一类”误差)。FDA 正在尝试在上市后监管期间采集和评价真实世界证据,以发现临床试验或其他上市前测试程序可能遗漏的风险证据。FDA 已启动多个真实世界数据采集计划,例如国家卫生技术评估系统(National Evaluation System for Health Technology,NEST)[56],如图5 所示,帮助提高真实世界证据质量,利用这些证据迅速发现新出现的安全信号并采取适当行动[57] ;或者借助MyStudies 应用程序,通过患者的移动设备促进真实世界证据采集,帮助制药企业设计新的医疗解决方案,同时遵守FDA 关于数据真实性、完整性和保密性的规定和指南[58]。通过分析所采集的真实世界证据,更快地识别确定范围更为广泛的安全信号,从而更快地为患者和医疗机构提供更精准的服务。

4. 强化内部AI 能力建设成为FDA 实现AI 前景、减少相关风险的关键
成功建立内部AI 体系的能力成为FDA 实现AI 前景并减少其风险的关键[3]。近年来,FDA 建立了强大的AI 嵌入式专业知识体系,通过重组数据运营并采用依赖于量身定制技术的数据基础设施,从医药产品监管机构逐渐发展为知识产出机构,成为AI 化的信息机构。
4.1 强化监管机构内部AI能力建设
美国联邦机构通常缺乏监管新型AI 产品或在内部构建AI 工具所需的技术能力,缺乏自行设计或评估算法系统的专业技术知识[59]。但FDA 属于例外,其投入了大量资源发展内部AI 能力,包括:投资于技术和数据基础设施;培养内部人才;开发并产出在技术层面可用、在法律和政策层面合规的AI 工具;制定全面和灵活的AI 战略,使FDA 能够从失败中战略性地学习和发展[60-61]。
作为美国公众健康的守护者,FDA 必须做好准备,使用与其所监管行业复杂程度相一致的工具和流程来应对未来的公共卫生突发事件。在与AI 相关的人力资源和专业知识体系的投资建设方面,从2017 年开始,FDA 陆续招聘数十名技术人员,包括云计算专家、AI 专家和工程师,帮助FDA 适应AI 技术的发展[1]。同年,FDA 创建“常驻企业家计划”(Entrepreneurs in Residence Program)[62]。FDA 下属机构器械与放射卫生中心(Center for Devices and Radiological Health,CDRH)希望通过“常驻企业家计划”,为监管机构的相关流程提供来自创业公司的新思路,这也是FDA 更广泛地切实推动创新举措的一部分。2021 年,美国卫生与公众服务部任命首位首席AI 官。同年首席AI 官办公室发布AI 战略,为在政府内部开发可信赖的AI 制定框架[63-64]。
FDA 充分发挥具备AI 专业知识的人才优势,通过试验监管沙盒和建立伙伴关系来提高监管机构内部AI 能力,开发制定灵活迭代的方法来监管基于AI 的医药产品[65]。监管沙盒为监管AI等快速发展的新兴技术提供了巨大的支持,使监管机构能够降低失败风险和相对安全地实施早期去风险项目,定义衡量成功的指标,同时迭代技术和基础设施,高效开展监管分析。FDA 尝试与制药企业、私营组织合作,将INFORMED 计划作为监管沙盒,聚焦于AI 驱动的肿瘤药物创新[35]。INFORMED 计划创建了一个独特的AI 监管沙盒,用于技术和组织资源的网络、构思和共享,为项目团队成功开发新型数据科学解决方案提供了所需工具。AI 监管沙盒使FDA 能够通过采取必要措施,在不危及公众安全的情况下开发功能更强的独立AI 工具。
依赖第三方开发人员构建AI工具的机构承担着允许受监管行业访问同一开发者的风险,从而可能损害相关工具。对此,FDA应采取措施,确保不依赖私营公司建立的“黑箱”专有AI 技术,避免不适当地有利于这些公司的监管目标。《算法政府:联邦行政机构中的AI》强烈鼓励各机构在AI 开发和维护方面构建内部专业知识体系[3]。
FDA 还通过与其他机构建立合作伙伴关系、鼓励公众参与相关科学挑战等增强FDA 内部技术能力。例如,2016 年FDA 与国家标准与技术研究所(National Institute of Standards and Technology,NIST) 建立合作关系,NIST 在制定评估AI 工具的风险和可信度水平的标准方面发挥了积极作用[66]。自2014年以来,FDA 通过向公众开放precisionFDA 挑战, 征求外部AI 专家的意见,帮助解决技术难题。该计划提供了安全的云端平台,参与者可以访问和共享数据集、数据管道和生物信息学工具;同时为社区成员提供了一个安全、独立的工作区,成员可自行决定在此使用相关生物信息学工具或数据,以便为其方法设定基准并推动监管科学的发展[67]。
如何加强技术数据基础设施建设,成为FDA 在内部能力建设领域面临的最大挑战。尽管FDA已经采取了多项举措来增加其AI工作可用的数据范围,并重点关注真实世界数据,但在将大数据转化为智能数据(smart data)方面仍存在诸多障碍[68]。FDA传统的上市前审评只基于制药企业提交的随机对照试验中的有限数据,这些数据涉及样本量小,采集时断时续,存储格式高度结构化。相比之下,FDA 的AI 上市后监测计划要求从各种来源采集数据,包括直接从患者处大量、持续采集真实世界数据。
4.2 AI 推动FDA 作为信息机构的崛起
现阶段FDA 通过FAERS和其他上市后监测相关数据库不断采集大量数据,未来是继续沿着以非结构化格式采集连续数据流的传统道路前进还是另辟蹊径,还有待进一步探讨。FDA 要想充分发挥作为信息机构的潜力,就必须开辟一条新的道路,即利用AI 技术,使用数据、内容和格式经过优化的量身定制的数据,以支持监管决策。其中不只是利用大数据,还需利用智能数据。FDA 应调整数据采集规程,首先采集结构化、量身定制的数据,而不只是局限于在建立基于NLP的工具后,从现有不良事件报告等非结构化文本中提取结构化数据。
FDA 目前对上市后监测的监管分析所采用的数据和AI 方法,完全依赖于应用于FAERS 等数据库的非结构化文本数据的NLP模型,这引发了相关人士对因果关系分析和数据互操作性的关切。尽管FAERS 试点计划在优先考虑使用者对上市后不良事件报告的审查以及发现药品和患者病情之间的新关系方面取得了一定的积极成果,但在尝试根据不具代表性的数据进行因果推断时,成功率较低。
一方面,FDA 并不要求证明产品与事件之间的因果关系,同时报告并不总是包含足够的细节用于正确评估事件[69]。另一方面,FAERS 报告还存在质量问题(数据重复)、完整性问题(数据缺失)和可靠性问题(数据未经核实)等[70]。
模型和模拟并非直接观察临床效果的替代物,而是因果关系证据的替代物。因此,模型和模拟可能比传统的替代物更不可靠。AI 的预测分析能力不能替代传统的因果推断,如果过度依赖计算机模型,可能会导致因果分析存在缺陷。FDA 目前对非结构化数据的依赖加剧了在因果推断方面的挑战。尽管精度在评估药物和不良健康状况之间的因果关系方面发挥着至关重要的作用,但在将非结构化数据转换为可操作的数据标记(例如药品名称或病症)的第一步就留下了不小的误差空间。由于很难合并包含非结构化数据的不同数据源,因此对非结构化数据的依赖使得数据互操作性的实现面临重大挑战。
在量身定制的数据上训练和使用AI 模型,能在一定程度上减轻FDA 在因果推断和数据互操作性方面的难度。结构化数据标记将使验证和执行数据质量、完整性和可靠性变得更加容易。《FDA技术现代化行动计划》(FDA'sTechnology Modernization Action Plan,TMAP)强调多个数据源之间以及FDA 与外部利益相关方之间的互操作性,同时强调结构化数据与非结构化数据并用[65]。
FDA 有能力将其采集的大部分数据转化为结构化数据。95%的FAERS 报告来自于可以按FDA 要求提交结构化数据的制造商。根据2018 年FDA 时任局长Scott Gottlieb 的披露,《真实世界数据企业方案》(Real World Data Enterprise Proposal)提出了1 亿美元的预算,用于扩展FDA采集的真实世界数据的数量和种类,协助上市后监管[71]。尽管在短期内需要更多的投入,但从长远来看,量身定制的数据为强大的监管分析用例提供了广阔的前景。继续保持或增加投入,有利于加强FDA 作为信息机构的监管作用。
5. 小结
AI 不仅为私营企业, 也为监管机构带来了巨大的发展前景。监管机构面临的问题并非在于是否采用AI,而是在哪些领域以及如何采用AI 来实现监管目的。FDA 已经有效地建立了内部AI 能力体系,积极主动勾勒出了一个以动态AI 为特色、为新型AI 医疗设备量身定制的监管框架,并在使用NLP 分析FAERS数据库中关于上市后不良安全事件数据方面取得具体进展。AI 技术的应用有利于加强上市后监管,加速监管范式转型。医药产品存在不确定性,与AI 有类似之处。作为医药产品的监管机构和率先探索AI 应用的政府机构,FDA针对AI 的监管经验、对数据驱动监管方法的进一步完善,以及对量身定制的数据的应用等,可为AI 在行政管理领域的未来应用场景提供示范,同时有望为其他机构提供有益的借鉴。
引用本文
姚立新.人工智能快速发展加速美国FDA 监管范式转型[J].中国食品药品监管.2025.2(253):22-35.

来源:中国食品药品监管杂志


