您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-11-01 17:17
刚刚,国家药监局器审中心发布《单纯疱疹病毒(HSV)核酸检测及分型试剂注册审查指导原则(征求意见稿)》,全文如下:
单纯疱疹病毒(HSV)核酸检测及分型试剂注册审查指导原则
(征求意见稿)
本指导原则旨在指导注册申请人对人单纯疱疹病毒核酸检测及分型试剂注册申报资料的准备及撰写,同时也为技术审评部门提供参考。
本指导原则是对单纯疱疹病毒核酸检测及分型试剂的一般要求,申请人应依据产品的具体特性确定其中内容是否适用。若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。
本指导原则是供注册申请人和技术审评人员使用的指导性文件,但不包括审评审批所涉及的行政事项,亦不作为法规强制执行,应在遵循相关法规的前提下使用本指导原则。如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究和验证资料。
本指导原则是在现行法规和标准体系以及当前认知水平下制定,随着法规和标准的不断完善,以及科学技术的不断发展,相关内容也将适时进行调整。
一、适用范围
本指导原则适用于利用荧光探针聚合酶链式反应(Real-time PCR)或其他类分子生物学方法的核酸检测技术,以特定的单纯疱疹病毒(Herpes simplex virus,HSV)1型和2型(HSV-1和HSV-2)基因序列为检测目标,对来源于人体样本中的单纯疱疹病毒DNA进行体外定性检测或分型检测,临床用于单纯疱疹病毒感染的辅助诊断。
单纯疱疹病毒属于人类疱疹病毒科α病毒亚科,为线性双链DNA 病毒,可分为单纯疱疹病毒-1型(HSV-1)和单纯疱疹病毒-2型(HSV-2),其核酸序列约有50%同源。HSV-1主要通过口-口接触传播造成口腔疱疹,但也可以引起生殖器疱疹,HSV-2属于性传播感染,可造成生殖器疱疹。感染HSV-2会加大罹患和传播HIV感染的风险。
临床用于检测单纯疱疹病毒的检测试剂,可根据不同的适应证选择适用的样本类型,主要样本类型为人泌尿生殖道拭子(如女性的宫颈或阴道拭子、男性的尿道拭子)。当应用于中枢神经系统感染时,可能涉及脑脊液样本。
对于采用其他方法学,或者采用其他样本类型的HSV核酸检测或分型试剂,可能部分要求不完全适用或本文所述内容不够全面,申请人应参照本指导原则,根据产品特性对适用部分进行评价,并补充其他的评价。
本指导原则适用于HSV核酸检测及分型试剂进行产品注册和变更注册的情形。本指导原则针对注册申报资料中的部分内容进行撰写,其他未尽事宜应当符合《关于公布体外诊断试剂注册申报资料要求和批准证明文件格式的公告》等相关法规要求。
二、注册审查要点
(一)监管信息
1. 产品名称及分类编码
产品名称应符合《体外诊断试剂注册与备案管理办法》及相关法规的要求,如单纯疱疹病毒-2型(HSV-2)核酸检测试剂盒(PCR-荧光探针法)。单纯疱疹病毒-1型和2型(HSV-1和HSV-2)核酸检测及分型试剂盒(PCR-荧光探针法)。按照《体外诊断试剂分类规则》,该产品按照第三类体外诊断试剂管理,分类编码为6840。
2. 申请人还需提交产品列表、关联文件、申报前与监管机构的联系情况和沟通记录及符合性声明等文件。
(二)综述资料
综述资料主要包括概述、产品描述、预期用途、申报产品上市历史及其他需说明的内容。应详细说明产品所采用的技术原理及检测流程,明确产品检测的靶基因及靶序列,需选择保守性和特异性相对较高的基因和靶序列,同时还应考虑扩增效率。其中预期用途需明确可检测的型别、靶基因、样本类型、适用人群、临床适应症等。HSV核酸检测试剂不建议对无症状人群进行检测。与已上市同类和/或前代产品的比较部分应着重包括方法学、检验原理、样本类型、采样部位及采样方式、检测靶基因、组成成分、内标、质控品,判读规则,不同HSV分型检出能力、预期用途、适用人群、分析性能和临床性能等方面。
(三)非临床资料
1. 产品技术要求及检验报告
按照《医疗器械产品技术要求编写指导原则》的要求编写产品技术要求,并提交三个不同批次符合产品技术要求的全项目检验报告。提交资料应符合《体外诊断试剂注册申报资料要求及说明》《医疗器械自检管理规定》等相关文件的要求。
2. 分析性能研究
注册申请人应当在原材料和生产工艺经过选择和确认、质量管理体系得到有效控制并且保证产品质量稳定的基础上,采用完整、确定的检测系统进行分析性能评估。
对于每项分析性能的评价都应包括具体研究目的、研究方法、试验方案、试验数据、统计分析等详细资料。有关分析性能验证的背景信息也应在申报资料中有所体现,包括实验地点、采用的试剂名称、规格和批号,仪器名称和型号,样本类型和来源等。分析性能评估的试验方法可以参考国际或国内有关体外诊断试剂性能评估的指导原则进行。
分析性能评估所用样本的基本信息均需明确,例如样本来源、样本类型、采集和处理方式、稀释方式、定值过程及数据等。研究中采用的HSV阳性样本,应采用科学合理的方法确定其型别、阴阳性和浓度水平,提交具体的试验资料。建议采用国际标准品建立校准曲线的方法确定研究样本的浓度。分析性能评估用样本一般应为真实样本,不建议采用质粒、假病毒等进行分析性能评估,如涉及稀释后检测,应采用与适用样本类型一致的阴性基质。对于各项性能中采用的样本,在下述各项性能研究资料中分别提供样本信息列表。检出限和包容性研究中所用样本应相互独立。
适用的不同样本类型应分别进行分析性能研究。如采用不同采样器、不同样本保存液、不同核酸提取与纯化等样本采集与处理方法,应分析这些差别的潜在影响,并进行针对性的分析性能验证,至少包括检出限、重复性、干扰性验证。
如试剂用于不同适用机型,需要在不同机型上分别进行性能评估。如申报产品包含不同的包装规格,需要对各包装规格进行分析或验证。
建议着重对以下分析性能进行研究。
2.1样本稳定性
应充分考虑实际使用过程中样本采集、处理、运输及保存等各个阶段的条件,对不同样本类型的样本稳定性分别进行研究。如有不同的样本采集处理方法(如不同采样拭子、采样过程,采集后未经处理的样本,加入不同裂解液/消化液的样本,灭活处理后的样本等)、不同的样本保存介质(样本保存液及保存容器)、不同的样本运输和保存条件(如涉及),则需分别完成稳定性研究。一般包括样本各种实际运输及保存条件下的保存期限验证。可冷冻保存的样本还应对冻融次数进行合理验证。
如核酸提取液可不立即进行检测,还需对核酸提取液的保存条件和保存时间进行研究。
2.2 适用的样本类型
结合产品的适用人群、预期用途及HSV分型,列明产品适用的样本类型。不同样本类型之间具有差异性,如申报产品包含多个样本类型,申请人应采用合理方法对不同样本类型分别进行全性能评估。
鉴于此类产品的样本采集方法不易标准化,并且对检测结果的准确性至关重要。因此,如产品采用不止一种样本采集方法(包括收集/保存介质),应针对不同的样本采集方法分别进行研究。
2.3企业参考品验证
根据主要原材料研究资料中的企业参考品设置情况,采用至少三批产品对企业参考品进行检验并提供详细的试验数据。
2.4准确度
可采用申报试剂与诊断准确度标准或已上市产品同时检测临床样本比较检测结果之间的一致性程度。研究应纳入一定数量的阴性和阳性样本,并注意包含一定数量的阳性判断值附近的样本和干扰样本。
不同的样本类型应分别进行准确度评价。
2.5 精密度
申请人应对精密度指标的评价标准做出合理要求,如标准差或变异系数的范围等。应对可能影响检测精密度的主要变量设计合理的试验方案进行精密度评价,包括运行、时间、操作者、仪器、试剂批次和地点等影响精密度的条件等。
精密度评价试验应包含核酸提取步骤。应设定合理的精密度评价周期,例如为期至少20天的检测。可采用临床样本或病毒株进行精密度评价,至少采用3个水平的临床样本进行精密度评价。具体要求如下:
阴性样本:待测物浓度低于最低检测限或为零浓度,阴性检出率应为100%(n≥20)。
临界阳性样本:待测物浓度略高于试剂的最低检测限,阳性检出率应大于95%(n≥20)。
中/强阳性样本:待测物浓度呈中度到强阳性,阳性检出率为100%,且Ct值的CV≤5%(n≥20)。
申请人应对检测数据进行统计分析,获得重复性、实验室内精密度、实验室间精密度、批间精密度等结果。建议对不同水平的HSV-1和HSV-2样本排列组合进行研究。
2.6检出限
应针对HSV-1和HSV-2分别进行最低检出限的建立和验证。使用已明确HSV分型和浓度/滴度的多例临床阳性样本/病毒株,采用与适用样本类型及样本保存液一致的阴性基质进行系列稀释,进行最低检测限的建立和验证。应采用合理方法确认HSV分型和浓度/滴度,提供详细的确认方法及结果。
2.6.1检出限确定
每个型别选取至少3份样本,系列稀释获得多个浓度梯度,每个浓度重复检测不少于3次,以100%可检出的最低浓度水平作为预设检测限。在此浓度附近制备若干浓度梯度样品,每个浓度至少重复检测20次,将具有95%阳性检出率的最低浓度作为最低检测限。也可采用适当的模型(如Probit 分析)和分析方法将具有95%阳性检出率的最低浓度水平作为确定的检出限。
2.6.2检出限验证
分别选择与2.6.1不同来源的HSV-1和HSV-2样本,至少各3例在检出限浓度水平进行验证,可采用阴性基质稀释到最低检测限浓度水平重复20次检测,应达到95%阳性检出率。
企业应能够提供用于最低检出限/定量限验证的病毒株的来源及浓度确认试验等信息。不同样本类型(如涉及)应分别评价最低检出限。
2.7包容性
2.7.1 采用生物信息学方法对产品检测的包容性进行分析,应覆盖已公布的单纯疱疹病毒核酸序列。
2.7.2 应选择具有时间和区域特征性的不同来源的HSV-1和HSV-2阳性临床样本或分型及浓度明确的HSV-1及HSV-2病毒株各不少于10例进行包容性验证,每种适用样本类型及适用的样本保存液均应进行验证,验证内容应包括重复性、检出限等。企业应能够提供用于包容性验证的样本来源、样本类型、型别及确认方法、浓度或滴度及其确定方法、实验数据等详细信息。对于检出限浓度附近样本未能检出的样本,应逐步提高浓度进行分析以确定其最低检出限。包容性验证所用临床样本或病毒株与最低检测限研究的样本不能重复使用。
2.8 分析特异性
2.8.1交叉反应
需验证多例人类基因组DNA和相关病原体的交叉反应。
用于交叉反应验证的病原体种类主要考虑下述的可能性:核酸序列具有同源性、易引起相同或相似临床症状、采样部位可能存在的其他微生物、其他常见病原体、临床需进行鉴别诊断的其他情形。
对于泌尿生殖道样本类型,建议对以下病原体进行交叉反应:单纯疱疹病毒1 型、单纯疱疹病毒2 型、乙型脑炎病毒、柯萨奇病毒、埃可病毒、肠道病毒(70、71),腮腺炎病毒、麻疹病毒、风疹病毒、淋巴细胞性脉络丛脑膜炎病毒、朊粒、水痘-带状疱疹病毒、EB病毒、巨细胞病毒、人疱疹病毒6 型、人类疱疹病毒7 型、人类疱疹病毒8 型、肠道病毒、人双埃可病毒、梅毒螺旋体、沙眼衣原体沙眼生物变种、沙眼衣原体性病淋巴肉芽肿变种、淋病奈瑟菌、杜克嗜血杆菌、解脲脲原体、阴道加特纳菌、厌氧菌、卷曲乳杆菌/惰性乳杆菌/克氏动弯杆菌/阴道陌生菌、普雷沃菌属/纤毛菌属、大肠埃希菌、无乳链球菌、干酪乳杆菌、阴道棒状杆菌、短小棒状杆菌、鲍曼不动杆菌、耻垢分枝杆菌、脆弱类杆菌、阴沟肠杆菌、粪肠球菌、金黄色葡萄球菌、表皮葡萄球菌/腐生葡萄球菌、甲型链球菌、乙型链球菌、白色念珠菌、光滑念珠菌、人型支原体、生殖支原体、刚地弓形虫、阴道毛滴虫。
对于脑脊液样本类型,建议对以下样本类型进行交叉反应研究:单纯疱疹病毒1 型、单纯疱疹病毒2 型、乙型脑炎病毒、柯萨奇病毒、埃可病毒、肠道病毒(70、71),腮腺炎病毒、麻疹病毒、风疹病毒、淋巴细胞性脉络丛脑膜炎病毒、朊粒、水痘-带状疱疹病毒、EB病毒、巨细胞病毒、人疱疹病毒6 型、人类疱疹病毒7 型、人类疱疹病毒8 型、肠道病毒、人双埃可病毒、脑膜炎奈瑟菌、肺炎链球菌、流感嗜血杆菌、大肠埃希菌、单核细胞增生李斯特菌、结核分枝杆菌、星状诺卡菌、苍白密螺旋体、无乳链球菌、肺炎克雷伯菌、无乳链球菌、化脓链球菌、琼氏不动杆菌、产酸克雷伯菌、新型隐球菌(新生/格特)、假丝酵母菌、曲霉菌。
建议在病毒和细菌感染的医学相关水平进行交叉反应的验证,通常,细菌感染的水平为106 CFU/mL或更高,病毒为105 PFU/mL或更高。
申请人应提供所有用于交叉反应验证的病毒和细菌的来源、阴阳性、种属/型别和浓度确认等试验资料。
对于某些难以培养或因为生物安全性无法培养的病原体,可采用病原体核酸样本进行交叉验证。应提供用于交叉反应验证的病原体核酸的来源、组成和浓度等信息,浓度可采用copies/mL单位表示。2.8.2 竞争性干扰
申请人应充分考虑临床上容易与HSV1或HSV2合并感染的病原体,在高浓度的情况下对低浓度(例如检出限浓度)HSV1或HSV2核酸检测的影响。建议申请人结合申报试剂的反应模式,使用一种最低检测限浓度附近的分析物和一种高浓度分析物评估竞争性干扰,此外对在低、中、高不同浓度条件下具有相同浓度水平的共感染菌株的竞争性干扰进行研究。竞争性干扰研究的病毒组合建议为同一反应体系内病毒、常见重症感染病毒及常见混合感染病毒。(如HSV-1和HSV-2的相互竞争性干扰,HIV对HSV-1和/或HSV-2的竞争性干扰和及其他可能共感染的病原体对HSV的竞争性干扰)。
2.8.3干扰试验
应针对不同样本类型,分别评价可能存在的干扰情况。建议在每种干扰物质的潜在最大浓度(“最差条件”),对包含临界阳性水平在内的样本分别进行干扰性研究。HSV-1和HSV-2需分别进行干扰性研究。需合理设置各干扰物的基质对照。对结果进行合理的统计分析,对比添加干扰物质前后的Ct值差异。潜在的干扰物质包括内源性、外源性、在样本采集和处理期间引入的物质及其他已报道的干扰物质。
泌尿生殖道拭子样本应至少选取全血(或血红蛋白)、白细胞、胆红素、宫颈粘液、粘蛋白、女性卫生用品、阴道常用药物(如避孕、抗真菌药物)、阴道栓剂、阴道润滑剂、阴道清洗剂等。
其他样本类型的干扰性研究应根据适用样本类型可能含有的干扰物质进行相应研究,需提供干扰物及浓度选择依据。
常用药物的干扰:四环素、阿奇霉素、螺旋霉素、罗红霉素、米诺环素、磺胺嘧啶、氢化可的松、地塞米松、阿昔洛韦、更昔洛韦、抗逆转录病毒药物等抗病毒药物。
2.9 HSV核酸DNA提取
在进行核酸检测之前,建议有核酸(DNA)提取/纯化步骤。该步骤的目的除最大量分离出目的DNA外,还应有相应的纯化作用,尽可能去除PCR抑制物。无论申报产品是否含有DNA分离/纯化的组分,企业都应对配合使用的所有核酸提取试剂进行提取核酸纯度、浓度、提取效率的研究,并与质量较好的核酸提取试剂进行平行比对。若产品适用两种或以上核酸提取试剂,则每一种核酸提取试剂均需配合检测试剂进行抗干扰、精密度和检出限的验证。
2.10 反应体系:
2.10.1样本采集和处理
2.10.1.1样本采集方式的选择
2.10.1.2样本采集时间点的选择:是否受病程、临床症状、用药情况等因素的影响。
2.10.1.3采样拭子及样本保存液的选择:对拭子头和拭子杆的材质要求。明确保存液或裂解液的成分、浓度、使用量的要求等。配套的不同拭子、不同保存液或裂解液至少需验证检出限、重复性及干扰性。
2.10.1.4样本处理方式的选择:研究产品适用的灭活方式,包括热灭活和化学灭活,研究内容包括胍盐的使用浓度及用量、样本用量。
2.10.2核酸提取和反应体系
研究确定最佳核酸提取和反应体系,包括核酸提取用的样本体积、洗脱体积和PCR加样体积、各种酶浓度、引物/探针浓度、dNTP浓度、阳离子浓度及反应各阶段温度、时间、循环数等。建议在保证核酸提取质量的情况下尽量扩大总反应体系和加样量,以提高检测灵敏度。
提交不同适用机型基线和阈值循环数的确定资料。
不同适用机型的反应条件如果有差异应分别详述,并提交验证资料。
2.11 提交携带污染研究资料。采用合理的检测顺序对多组高浓度阳性样本和阴性样本交替检测评估携带污染。
3. 稳定性研究资料
稳定性研究资料主要涉及两部分内容,申报试剂的稳定性和适用样本的稳定性研究。前者主要包括实时稳定性(有效期)、运输稳定性、开瓶稳定性及冻融次数限制等研究,申请人可根据实际需要选择合理的稳定性研究方案。稳定性研究资料应包括研究方法的确定依据、具体的实施方案、详细的研究数据以及结论。对于实时稳定性研究,应提供至少三批样品在实际储存条件下保存至成品有效期后的研究资料。
4. 阳性判断值研究资料
申请人应考虑不同地理区域流行病学背景以及人口统计学特征(包括性别、地域、种族等因素)的差异,选择具有代表性的样本建立阳性判断值,注意应纳入一定数量的弱阳性和高阴性水平样本。如采用其他研究方法,应说明其合理性。
对于荧光实时PCR方法即为用于结果判读的Ct值和/或核酸浓度的确定资料,包括确定基线阈值、阈值循环数(Ct)、核酸浓度(如适用)的研究资料等。如判定值存在灰区,应提供灰区的确认资料。
另外,建议申请人考虑建立阳性判断值时使用的受试者样本对于目标人群的代表性,通过临床评价进一步验证和确认阳性判断值的准确性。
提供内标检测结果范围的确定方法和研究资料。
5. 其他资料
5.1 主要原材料的研究资料
此类产品的主要原材料包括引物、探针、酶、dNTP、核酸分离/纯化组分(如有)、企业参考品、对照品、质控品等。应提供主要原材料的来源、选择、制备方法的研究资料,质量分析证书,主要原材料质量标准的制定和检验资料。如主要原材料为企业自制,应提供其详细的制备、鉴定和质量控制过程,其制备工艺必须相对稳定;如主要原材料源于外购,应提供资料包括:选择该原材料的依据及对比筛选试验资料、选定的供应商名称,供应商提供的质量标准、原材料检验报告(质量证书),以及该原材料到货后的入厂检验报告,供应商应为原材料的生产商,不得随意更换。申请人应对各主要原材料均明确质量控制标准。
5.1.1核酸分离/纯化组分(如有)的主要组成、原理介绍及相关的验证资料。
5.1.2引物和探针
应详述引物和探针的设计原则,包括对包容性和特异性的考虑情况。提供引物、探针核酸序列、模板核酸序列及两者的对应情况。建议每种病毒设计两套或多套引物、探针以供筛选,针对所有预期适用的基因型别进行检出能力和特异性(如交叉反应)的评价,选择最佳组合,并提交筛选的研究数据。引物、探针的质量标准应至少包括序列准确性、纯度、浓度、探针标记的荧光素及功能性实验等。申请人应提供验证资料或合成机构出具的质检证明,如PAGE电泳结果或高效液相色谱法(HPLC)分析图谱等。
5.1.3脱氧三磷酸核苷(dNTPs)
包括:dATP、dUTP、dGTP、dCTP和dTTP;应提交对其纯度、浓度、保存稳定性等的验证资料。
5.1.4酶
需要的酶主要包括DNA聚合酶、尿嘧啶DNA糖基化酶(如有)等,应分别对酶活性、功能性等进行评价和验证。
DNA聚合酶,应具有DNA聚合酶活性,无核酸内切酶活性,具热稳定性,如:94℃保温1小时后仍保持50%活性。根据酶的特性明确其外切酶活性。
尿嘧啶DNA糖基化酶(UDG/UNG),具有水解尿嘧啶糖苷键的活性,无核酸外切酶及核酸内切酶活性。应对酶活性及热稳定性进行合理验证。
5.1.5核酸类检测试剂的包装材料和耗材应无脱氧核糖核酸酶(DNase)和核糖核酸酶(RNase)污染。
5.1.6企业参考品:
如适用,应提交企业参考品的原料来源、组成、毒株特性(如名称、型别、浓度)、阴阳性及浓度/滴度确认方法或试剂等相关验证资料(溯源及定值资料等信息)。企业参考品的核酸性质应与产品预期检测的靶物质一致。企业参考品的基质与临床样本收集/保存介质一致。建议采用灭活病毒的临床样本或病毒培养物建立参考品,不宜使用质粒、假病毒、基因组提取/纯化物等。
企业参考品的项目应包括:阳性参考品、阴性参考品、最低检测限参考品、精密度参考品等。
阳性参考品,无论试剂盒能否进行HSV分型,阳性参考品均应针对所有适用的HSV分型分别设置, 并设置不同滴度水平。
阴性参考品应考虑分析特异性的评价,应纳入正常临床样本、含干扰因素的样本、不在试剂盒检测范围内的HSV分型、易引发相似症状的、易共感染的其他病原体阳性样品。
最低检测限参考品:申请人应明确检测限参考品中病毒核酸浓度的确定方法,明确检测限参考品中病毒核酸浓度的确定依据,检测限参考品中HSV核酸浓度应为申报产品检测限浓度或略高于检测限浓度。
精密度参考品应至少包含两个浓度水平,其中包含弱阳性水平。
5.1.7 内对照(内标)、质控品
内对照(内标)设置应合理,阴/阳性质控品宜采用混合临床样本或病毒株或假病毒制备。
内标以对管内抑制可能造成的假阴性结果进行质控。申请人应对内标的引物、探针和模板的浓度做精确验证,保证内标荧光通道呈明显的阳性曲线,不应对目的基因的检测造成竞争性抑制而导致假阴性,对内标的Ct应有明确的范围要求。
阴/阳性质控品应参与样本核酸的平行提取,以对整个PCR反应过程、试剂/设备、交叉污染等环节进行合理质量控制。企业应对各种质控品的Ct做出明确的范围要求。
5.2 主要生产工艺及反应体系的研究资料
基本生产工艺主要包括:配制工作液、半成品检定、分装和包装。配制工作液的各种原材料及其配比应符合要求,原材料应混合均匀,配制过程应对pH等关键参数进行有效控制。
生产工艺研究的资料应能对反应体系涉及到的基本内容,如样本类型、样本用量、试剂用量、反应条件、校准方法、质控方法、稳定性和有效期,提供确切的依据,主要包括以下内容:
5.2.1主要生产工艺介绍,可以图表方式表示。
5.2.2 DNA提取纯化方法优化,建议包含纯化步骤,内标、质控品均应全程参与提取纯化。
5.2.3确定最佳PCR反应体系的研究资料,包括酶浓度、引物/探针浓度、dNTP浓度、阳离子浓度、样本量、加样量及反应体积等。
5.2.4确定PCR各阶段温度、时间及循环数的研究资料。
5.2.5对于基线阈值和阈值循环数(Ct)确定的研究资料。
5.2.6不同适用机型的反应条件的对比分析,如果有差异应分别详述。
(四)临床试验
临床试验的开展、方案的制定以及报告的撰写等均应符合相关法规、《医疗器械临床试验质量管理规范》及《体外诊断试剂临床试验技术指导原则》的要求。下面仅说明该类产品临床试验中应关注的重点问题。
1. 研究方法
对于已有同类产品上市的试剂的临床研究,选择境内已批准上市、临床普遍认为质量较好的同类产品作为对比试剂,采用试验体外诊断试剂与之进行对比试验研究,评价本产品的临床性能。
2.适用人群
临床试验受试者应包括各种可能接受单纯疱疹病毒感染检查的人群,具有单纯疱疹病毒感染疑似症状的患者,如生殖器疱疹、丘疱疹、疼痛性丘疹、有不安全性接触史的人群。男性和女性受试者应尽量均匀分布。
3.临床试验病例数
适当的样本量是保证试验体外诊断试剂临床性能得到准确评价的必要条件。临床试验样本量应满足统计学要求,可采用适当的统计学方法进行估算。根据相应临床试验设计,可选择阴性符合率/特异度和阳性符合率/灵敏度,分别估算最低阴性样本例数和阳性样本例数。
如临床试验采用试验体外诊断试剂与已上市同类产品进行比对的试验设计,可采用单组目标值法样本量公式估算最低样本量。
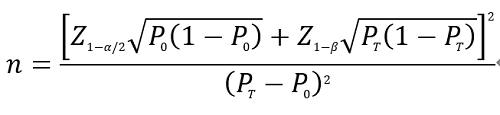
公式中,n为样本量;Z1-α/2、Z1-β为显著性水平和把握度的标准正态分布的分位数,P0为评价指标的临床可接受标准,PT为试验体外诊断试剂评价指标预期值。
基于现有认知,评价指标的临床可接受标准(P0)即HSV-1/HSV-2核酸试剂阳性符合率和阴性符合率原则上建议不低于95%。当评价指标P接近100%时,上述样本量估算方法可能不适用,应考虑选择更加适宜的方法进行样本量估算,如精确概率法等。如临床试验研究有更合理的样本量估算方式,在说明其合理性后亦可采用。
样本量估算过程中需要考虑临床试验中病例的脱落剔除率,一般而言,病例脱落剔除率不应高于10%。此外,临床试验应纳入一定数量的弱阳性样本。
如申报产品具有多种适用的样本类型,不同样本类型之间存在较大差异,例如:分析性能不同、临床性能不同、适用人群不同、适应症不同、阳性判断值不同等,则可能需要分别进行临床试验设计,包括分别进行样本量估算和统计学分析。
如申请产品适用于脑炎/脑膜炎等其他相关适应证,应根据产品预期用途,综合考虑产品风险进行科学的临床试验设计。
4.临床研究单位的选择
应选择不少于3家(含3家)临床试验机构,按照相关法规、指导原则的要求开展临床试验。临床试验机构的选择应尽量考虑试验体外诊断试剂的特点和预期用途,综合流行病学背景,受试者的选择具有一定的地域代表性。且临床试验机构应具有分子生物学方法检测的优势,实验操作人员应有足够的时间熟悉检测系统的各环节,熟悉评价方案。
5. 临床试验数据统计分析
应选择合适的统计方法对临床试验结果进行统计分析,对于试验体外诊断试剂与对比试剂/方法的一致性评价,一般选择2×2表的形式总结两种试剂/方法的检测结果,并据此计算阳性符合率/灵敏度、阴性符合率/特异度、总符合率、Kappa值等指标及其置信区间。
对于不一致样本,应进行原因分析。如临床试验方案规定采用其他方法进行确认,则确认结果不应纳入统计分析。
6.伦理学要求
临床试验必须符合赫尔辛基宣言的伦理学准则,必须获得临床试验机构伦理委员会的同意。研究者应充分考虑临床试验用样本的获得和试验结果对受试者的风险性,应提交伦理委员会的审查意见。
7.质量控制
临床试验开始前,建议进行临床试验的培训,以熟悉并掌握相关试验方法的操作、仪器、技术性能等,最大限度控制试验误差。整个试验过程都应处于有效的质量控制下,最大限度保证试验数据的准确性及可重复性。
8. 临床试验方案
体外诊断试剂临床试验应按照同一临床试验方案在多家临床试验机构开展,且保证在整个临床试验过程中遵循预定的方案,不可随意改动。整个试验过程应在临床试验机构的实验室内并由该实验室的技术人员操作完成,申报单位的技术人员除进行必要的技术指导外,不得随意干涉实验进程。
试验方案应确定严格的入选/排除标准,任何已入选的样本被排除出临床试验都应记录在案并明确说明原因。在试验操作过程和结果判定时,应采用盲法以保证试验结果的客观性。各临床试验机构选用的对比试剂/方法应保持一致,以便进行合理的统计学分析。另外,应考虑试验体外诊断试剂样本类型与对比试剂/方法样本类型的可比性;如有差异,需提供充分的验证。
9. 临床试验报告
应对试验的整体设计及各个关键点给予清晰、完整的阐述,应该对整个临床试验实施过程、结果分析、结论等进行条理分明的描述,并应包括必要的数据和统计分析方法。
(五)产品说明书
产品说明书格式应符合《体外诊断试剂说明书编写指导原则》的要求。产品说明书中技术内容均应与注册申报资料中的相关研究结果保持一致,如某些内容引用自参考文献,应以规范格式进行标注,并单独列明文献的相关信息。
HSV病毒核酸检测及分型试剂说明书编写应重点关注以下内容。
1.【预期用途】
1.1试剂盒用于体外定性检测人体样本(如男性尿道拭子、女性宫颈或阴道拭子)中的HSV-1和HSV-2 核酸(DNA),如可分型则需明确。适用样本类型应依据申报产品的分析性能评估和临床研究情况进行确认。
1.2结合临床和其他诊断指标,可用于单纯疱疹病毒感染的辅助诊断。
1.3 目标人群:各种可能接受单纯疱疹病毒核酸检查的人群。例如生殖器疱疹、丘疱疹、疼痛性丘疹、有不安全性接触史的人群。
1.4 临床背景信息:简要描述病原体生物学特征及致病性,流行病学特征、潜伏期、易感人群、感染后临床表现及相关疾病等。简要介绍现有HSV临床或实验室诊断方法。
2.【检验原理】
简述核酸提取纯化及试剂盒检测原理,明确产品能够检测的目标基因信息及序列特征,明确内标基因名称及其作用,明确引物探针设计及探针标记荧光信息如采用了防污染措施,进行简要描述。
3.【主要组成成分】
明确试剂盒中各组分及具体成分,明确需要但未提供的材料,如核酸提取试剂、样本采集器械、病毒保存液等的产品名称、生产厂家、货号及注册证号或备案号等信息。
4.【样本要求】
4.1样本收集要求:针对不同的样本类型分别详细描述样本采集和处理方式,包括采样步骤、灭活方式、采样体积、离心要求等。如有临床公认推荐的采样要求,应遵循,并引用相应的技术规范或指南。
4.2明确经验证的配套使用的采样管、拭子、保存液等信息。
4.3样本处理、运送及保存:根据研究资料,明确对样本核酸提取前的预处理、运送条件、保存条件及期限等信息。根据适用的情形,针对采样后放入保存液前的干燥拭子、加入样本保存液后核酸提取前、核酸提取后样本等不同阶段的处理方式、保存稳定性及运送条件等。如声称样本可冷冻,需明确冻融次数如有需要应对高于检测范围的样本稀释方法进行规定。
5.【检验方法】
详细说明试验操作的各个步骤。
明确核酸提取用的样本体积、洗脱体积和PCR加样体积,阴阳性质控品与待测样本同步进行核酸提取操作。明确各适用机型的反应参数设置,基线、循环阈值(Ct值)的选择方法,以及各检测靶标对应的荧光通道。明确内标、质控的检测结果Ct值范围。CtCt
6.【检验结果的解释】
结合阳性对照、阴性对照、内对照(内标)以及目标基因的检测结果,通过扩增曲线和Ct值进行阴阳性的判断,详述所有可能出现的结果组合及相应的解释Ct。如存在灰区,应同时说明对灰区的处理方式,包括在何种情况下需要进行重复检测,重复检测的方法,对样本可能采取的优化条件(如采集要求或采集方法)等。
7.【检验方法局限性】
7.1本检测结果仅供临床参考,如需确诊病例请结合临床症状及其他检测手段。
本试剂盒的检测结果应结合患者的症状/体征、病史、其他实验室诊断结果等情况进行综合分析以及解释,不得作为患者临床诊治或管理的唯一依据。
7.2 HSV DNA检测阴性不能排除HSV 感染。
7.3导致假阴性/假阳性结果的可能性分析:
7.3.1不合理的样本采集、处理、运输及保存条件,样本中目标物滴度过低。
7.3.2 HSV病毒核酸目标基因序列的变异或其他原因导致的序列改变。
7.3.3同一患者不同时间、不同部位或者多次采集样本会降低假阴性结果的可能性。
7.3.4未经验证的其他干扰,如内源性或外源引入样本的物质。
7.3.5样本间的交叉污染。
7.3.6未经验证的其他交叉反应物质。
8.【产品性能指标】详述以下性能指标:
描述产品性能,包括以下内容:
8.1 国家标准品(如有)和企业参考品的符合率。
8.2 检出限:说明不同类型样本的最低检测限,简要介绍最低检出限的确定方法以及验证最低检出限所采用的菌株分型。如HSV-1和HSV-2最低检测限不同,应分别列出。
8.2精密度:说明不同类型样本的重复性和重现性评价结果。
8.3分析特异性:包括交叉反应和干扰物质。
8.3.1可能产生交叉反应的其他病原体的验证情况,建议以列表的方式描述病原体名称、型别、浓度等信息。
8.3.2样本中常见干扰物质对检测结果的影响,应注明可接受的最高限值。
8.4临床试验:简要介绍试验方法、受试者及样本、试验结果和结论等。
9.【注意事项】应至少包括以下内容:
9.1 有关人源组分(如有)的警告,如:试剂盒内校准品、质控品或其他可能含有人源物质的组分,虽已通过乙型肝炎表面抗原(HbsAg)、人类免疫缺陷病毒抗体(抗-HIV1/2)、丙型肝炎抗体(抗-HCV)等项目的检测为阴性,但截至目前,没有任何一项检测可以确保绝对安全,故仍应将这些组分作为潜在传染源对待。
9.2 临床实验室应严格按照《临床基因扩增实验室工作规范》配备设备及操作人员,应严格按照说明书要求进行操作。
三、参考文献
[1]体外诊断试剂注册与备案管理办法[Z].
[2]关于公布体外诊断试剂注册申报资料要求和批准证明文件格式的公告[Z].
[3] 体外诊断试剂临床试验技术指导原则[Z].
[4]体外诊断试剂说明书编写指导原则[Z].
[5] WS/T 236-2017 《生殖器疱疹诊断》[S]
[6] WHO. Laboratory diagnosis of sexually transmitted infections, including human immunodeficiency virus. [Z].
[7] WHO. Guidelines for The Management of Symptomatic Sexually Transmitted Infections.[Z].
[8]CDC. Sexually Transmitted Infections Treatment Guidelines, 2021 [Z].

来源:国家药监局


