继今年10月13日欧盟医疗器械委员会提出医美器械监管提案之后,到现在已经过去了快两个月,就在12月1日,欧盟医美器械的监管法案正式发布,也就意味着,医美器械在欧盟需要开始做认证了。


这个法案规定了以下两部分内容:
1. 无医疗用途的Annex XVI产品的通用规范:规定了医美器械风险管理(包括标签和使用说明)和临床评估方面的要求;
2. 无医疗目的某些有源产品类别的重新分类规则:对某些无医疗目的的有源产品(如IPL、吸脂和脑刺激产品)进行重新分类。
生效日期和强制实施日期
重新分类的要求将于2022年12月22日开始生效。
通用规范的要求将于2022年12月22日开始生效,2023年6月22日强制实施,但是Article 2的第3条的要求(即此类器械的遗留器械的要求)将从2022年12月22日开始强制实施。
该法案适用于MDR Annex XVI产品范围中包含的以下六个特定产品组:
1) 隐形眼镜或其他拟引入眼睛或进入眼睛的物品。示例包括非处方彩色隐形眼镜。
2) 旨在通过外科侵入手段将全部或部分引入人体的产品,目的是改变人体部位的解剖结构或固定,但纹身产品和穿孔除外。
3) 用于面部或其他皮下、粘膜下或皮内注射或其他引入的物质、物质组合或物品,不包括用于纹身的物质。示例包括真皮填充物。
4) 用于减少、移除或破坏脂肪组织的设备,如吸脂、脂肪分解或脂肪成形设备。
5) 用于人体的高强度电磁辐射(如红外线、可见光和紫外线)发射设备。例如,用于纹身或脱毛的激光和强脉冲光(IPL)设备。
6) 用于大脑刺激的设备,该设备应用电流或磁场或电磁场穿透颅骨以改变大脑中的神经元活动。
医美器械的重新分类
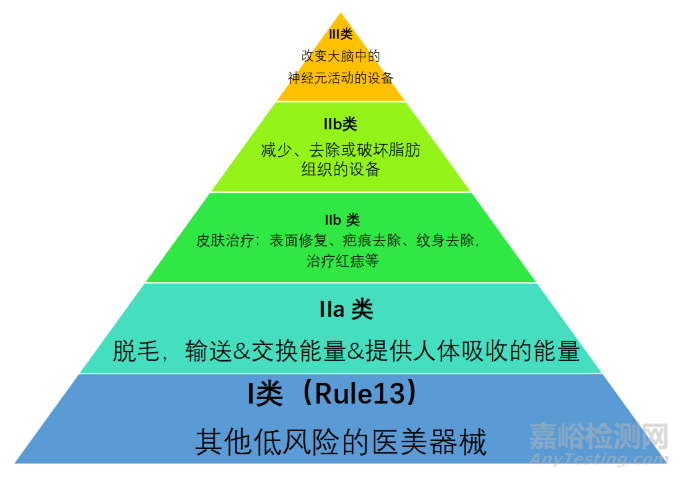
通常,以上产品组中的低风险设备被重新分类为I类。
分类规则为MDR法规中的Rule 13 (All other active devices are classified as class I
但是,根据可用于人体的高强度电磁辐射设备的现有科学证据,如激光和强脉冲光('IPL')设备,使用此类设备可能会产生副作用,例如表面烧伤、炎症、疼痛、色素沉着变化、红斑、增生性瘢痕和水泡。副作用通常表现为短暂的,例如炎症,但也有重要和持久的影响,如皮肤色素沉着变化。
根据风险大小,将用于人体的高强度电磁辐射设备分为IIa, IIb或III类:
用于人体脱毛的无预期医疗用途的高强度电磁辐射发射设备,如向人体输送能量或与人体交换能量或提供人体吸收的能量的激光和IPL设备,应归类为IIa类。
用于人体皮肤治疗的无预期医疗用途的高强度电磁辐射发射设备,应归类为IIb类,如用于皮肤表面修复、疤痕去除、纹身去除,或用于治疗红痣、血管瘤的激光或IPL设备。
用于减少、去除或破坏脂肪组织的设备,如抽脂、射频脂解、超声脂解、冷冻脂解、激光脂解、红外和电刺激脂解、声学冲击波治疗或脂肪成形术设备,应归类为IIb类。
应用电流或磁场或电磁场穿透颅骨,以改变大脑中的神经元活动的用于大脑刺激的设备,例如用于经颅磁刺激或经颅电刺激的设备,应归类为III类。
符合性评估途径
根据本法规的重新分类,根据法规(EU)2017/745 的Article 52,公告机构将参与相关产品的符合性评估,以评估和确认在相关的一般安全和性能要求中,产品达到预期性能,并且产品所带来的风险已尽可能消除或降低。
此类器械的遗留器械的要求
本法规适用的产品,以及由公告机构根据指令93/42/EEC颁发的证书所涵盖的产品,可在2028年6月22日或2025年6月22日之前(即使证书到期),投放市场或投入使用,前提是满足以下条件:
(a)该产品在2023年6月22日之前已经在欧盟合法销售,并继续符合指令93/42/EEC的要求,但如果证书在2021年5月26日之后到期,则需要由公告机构颁发的有效证书覆盖;
(b)产品的设计和预期用途没有重大变化;
(c)在公告机构根据指令93/42/EEC颁发的证书到期日之后,制造商需要与公告机构签署相关书面协议,确保符合上述(a)和(b)点要求。
可在2028年6月22日之前投入市场或投入使用的产品,除了满足上述(a)~(c)条件之外,还需符合以下条件:
制造商打算或正在对其进行临床研究以生成临床评估的临床数据,以确认其符合法规(EU)2017/745附录I中规定的相关一般安全性和性能要求以及本法规中规定的通用规范的产品,并符合公告机构必须参与的符合性评估。
从2024年6月22日至2024年12月22日,符合该规定条件的产品能投放市场或投入使用的前提是,赞助商已收到相关成员国根据法规(EU)2017/745 Article 70的第(1)或(3)条发出的通知,确认产品临床研究申请已完成,且临床研究属于法规(EU)2017/745的范围。
从2024年12月23日至2026年6月22日,符合该规定条件的产品只能在赞助商已开始临床研究的情况下投放市场或投入使用。
从2026年6月23日至2028年6月22日,符合该规定条件的产品只能在公告机构和制造商签署了执行符合性评估的书面协议(written agreement for the performance of the conformity assessment)的情况下投放市场或投入使用。
可在2025年6月22日之前投入市场或投入使用的产品,除了满足上述(a)~(c)条件之外,还需符合以下条件:
如果制造商不打算对其进行临床调查,但公告机构必须参与其符合性评估。
从2023年9月22日至2025年6月22日,符合该规定条件的产品只能在公告机构和制造商签署了执行符合性评估的书面协议的情况下投放市场或投入使用。