随着国内IVD技术进步及国家相关政策政策变化的推动,越来越多IVD公司积极开拓海外业务,布局全球市场。全球医疗器械的主要市场集中在美国、欧洲、日本、中东、阿拉伯地区和中国,其中美国的医疗器械生产和消费都占到几乎全球的50%左右。中国医疗器械产品主要出口市场包含美国、欧盟、英国、澳大利亚、加拿大、韩国、日本等。本文对各主要国家及地区医疗器械注册要求及流程进行了汇总。
美国 FDA
市场价值:2021年,全球医械市场规模达5000多亿美元。据媒体报道,美国医械市场占据了全球41%的市场份额。
FDA将医疗器械分为三类(Ⅰ、Ⅱ、Ⅲ),Ⅲ类风险管控等级高于Ⅰ、Ⅱ类,这一点和国内保持一致。FDA将每一种医疗器械都明确规定其产品分类和管理要求,目前FDA对大约1700多种医疗器械产品进行了分类涉及16个不同的版块。任何一种医疗器械想要进入美国市场,必须首先弄清申请上市产品分类和管理要求。
步骤1:器械分类
如果想获得 FDA 的正式器械确定或分类,可向FDA提交 513(g) 请求。但一般需要支付相应的费用。医疗器械依据其风险的程度,分为以下3类:
Ⅰ类-低等风险(监管控制类型:基本控制)大部分并不是所有,一般不需要510(K);
Ⅱ类-中等风险(监管控制类型:基本控制以及特殊控制)大部分并不是所有,一般需要510(K);
Ⅲ类-高等风险(监管控制类型:基本控制以及上市前批准)。
步骤2:选择正确的路径递交
器械分类确定之后,需要选择相应法规要求下的上市前递交。
常见的上市前递交类型包括:
• 510(k)(上市前通知)
• PMA(上市前批准)
• De Novo(自动III类指定的评价)
• HDE (人道主义器械豁免)
一些Ⅰ类以及大部分Ⅱ类器械要求以510(k)的方式递交。在510(k)递交过程中,申请者必须证明新的器械与对比器械在预期用途,技术特征以及性能测试方面实质等同。一些Ⅰ类和Ⅱ类器械可以豁免510(k),如果他们在21 CFR 862-892.9所述的豁免范围之内。这些豁免被列在21 CFR的分类规则中,也被汇集在医疗器械豁免文件中。
大部分Ⅲ类器械要求的递交方式为PMA。PMA为严格程度较高的上市前递交类型。在FDA批准PMA之前,申请者必须提供有效的科学证据,以证明器械预期用途的安全性以及有效性。
De Novo为没有有效对比的新器械提供一种方式,如果这种新器械满足特定标准,可以被分为Ⅰ或Ⅱ。
HDE为Ⅲ类器械提供了一种监管路径,这类器械预期对罕见疾病或状况的患者是有益的。器械有资格成为人道主义豁免器械,申请者必须获得人道主义使用器械(HUD)的指定,可通过向FDA的孤儿产品开发办公室Office of Orphan Products Development (OOPD)递交申请。
步骤3:准备材料
在选择正确的上市前递交类型之后,必须准备该递交类型所需的适当的资料。本部分将介绍在准备上市前递交时需要用到的有用资源以及考虑的信息。FDA开发一些帮助申请者准备上市前递交的资源类型,包括:
器械建议(Device Advice)—综合基于FDA上的网页法规协助
510(k)的准备,参考:Premarket Notification 510(K)
PMA的准备,参考:Premarket Approval (PMA)
CDRH学习(CDRH Learn) —基于视频的教学模块,研讨会和录制的包括各种政策和指导力度的网络研讨会
CDRH递交前程序 —未来上市前提交申请可能要求FDA通过这个程序进行反馈
准备上市前递交时需要考虑的信息:
设计控制:所有II类以及III类器械根据质量管理体系(21 CFR 820.30)中对设计控制的要求进行设计。一些I类器械可豁免设计控制。
非临床测试:器械上市要求的测试以及信息类型是通过器械的分类,作用机制,技术特征,以及标签来确定的。医疗器械上市前递交实施的非临床测试必须符合21 CFR 58中的良好试验管理规范(GLPs)
临床证据:PMAs,HDEs 以及部分 510(k)s 和 De Novos 要求有临床证据。在早起的临床研究开始之前,研究申请者需要得到FDA器械临床研究豁免(IDE)的批准。这项研究也需要得到伦理审查委员会(IRB)的批准。临床研究必须符合所有的适用的器械临床研究豁免(IDE)法规以及良好试验管理规范(GLPs)。
标签:器械的标签必须依据标签法规书写,且需要包含在上市前递交的资料中。
步骤4:递交资料
提交给FDA,并在FDA的工作人员审查过程中保持联系。
用户费用:在510(k)或PMA递交时,需要一定的用户费用
电子副本(eCopy):上市前递交必须包含以光盘(CD)、数字视频光盘(DVD),或闪存驱动器方式形成的电子副本。
行政备案审查:在上市前递交接收之后,FDA进行行政审查,评估递交是否是足够完整的,以接收实质性审查。
审查互动(Interactive Review):当递交的资料处于正在审查中时,FDA将和申请者保持联系以增加审查过程中的效率。
步骤5:完成登记
器械设备必须在FDA对其生产的企业进行登记,并对其器械进行列名。如果一个器械在上市前需要上市前清关(premarket clearance)或上市前批准(premarket approval),器械厂商在登记和列名之前必须等到它获得FDA的清关或批准。器械企业登记、登记号的分配或医疗器械的列名,都不意味着FDA对其企业或其产品的清关或批准。
欧洲CE
市场价值:根据MedTec Europe统计,2022欧洲医疗器械市场规模约为1350亿欧元,约占全球市场的27%,是仅次于美国的第二大医疗器械市场。
自2021年5月起,医疗器械制造商必须遵守欧盟医疗器械法规2017/745而不是医疗器械指令93/42/EEC,才能获得CE标志批准。因此,医疗器械必须根据MDR进行分类。但是,一些分类规则保持不变,因此请随意检查以下MDD分类路线。
如果您是制造商并且希望将您的医疗设备投放到欧盟市场,您需要确保其符合欧盟委员会制定的特定欧洲指令。在这种情况下,重要的是医疗器械指令(MDD):AIMDD90/385/EEC;MDD93/42/EEC;IVDMDD98/79/EC。为了证明您的设备符合这些CE指令的基本要求,您需要在其上贴上CE标志。您的产品需要通过CE认证标记过程。后者的方向取决于您的医疗器械类别和您选择的合格评定途径。您的医疗设备的具体特性将决定其类别,以及对患者的风险程度。例如,预期用途、侵入性以及局部与全身效应等特征。
根据欧洲框架,医疗器械分为四类:I类、IIa类、IIb类和III类。III类医疗器械的风险最高。今天,由于新监管系统的更严格规则,许多设备的类别发生了变化。之前他们会被归入IIa或IIb类,但现在他们将被归入III类。如果您的医疗设备属于I类以外的任何其他类别,您必须向认证机构提供证明,证明您的产品符合相应CE指令的基本要求。
第一类医疗器械CE
医疗器械类I的风险最低。此类设备的制造商可以选择三种可能的CE标记途径中的一种。在这方面,他们应该考虑以下几点:如果医疗器械是否是无菌的,例如个人防护套件;医疗器械是否具有测量功能,如听诊器;如果它不是无菌的,也不是测量的,例如矫正眼镜。注意:如果您的产品是I类产品,并且它不是无菌或测量设备,那么您只需对其进行自我认证,并通过书面声明正式声明其符合MDD的适用要求。如果它是无菌或测量医疗设备,那么您将需要认证机构评估。

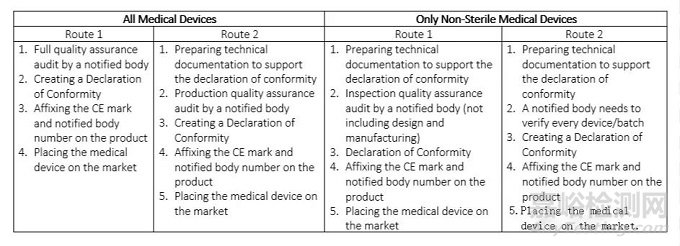
表1:I类医疗器械的CE认证标志路线
IIa类医疗器械CE
IIa类医疗器械可能是手术手套、助听器、诊断超声机等。它们通常构成中低风险。患者应短期使用,不超过30天。如果您是IIa类医疗设备的制造商,您必须支持您的声明符合认证机构评估。只有这样,您才能将您的产品投放市场。有四种可能的途径可以让您的产品获得CE标志,根据产品的类型(即是否无菌)分为两组。
表2.IIa类医疗器械的CE标志路线
IIb类医疗器械CE
在这里,我们可以包括医疗设备,例如长期矫正隐形眼镜、手术激光、除颤器等。它们是中高风险设备,患者可能会使用它们超过30天。如果您的产品属于IIb类,类似于IIa类的程序,您将需要指定机构来评估您的技术文档是否符合医疗器械指令。特定CE标志路线的选择将再次取决于您的产品类型。
表3.IIb类医疗器械的CE标志路线
第三类医疗器械CE
在该类别中,所有医疗设备都具有最高的风险,并且需要在其生命周期内进行永久监控。有专门的机构负责对产品进行监测。例如,此类装置是心血管导管、动脉瘤夹、髋关节植入物、人工心脏瓣膜等。在这里,以及在II类中,医疗器械的合格评定可能包括对技术文件的审核和质量体系/产品检查,并侧重于器械设计和生产的一个或多个方面。
表4三类医疗器械的CE标志路线
英国UKCA
英国脱欧后,UKCA 标志于 2021 年 1 月在英国大不列颠 (Great Britain) 正式生效。一些分类的医疗器械可能需要持有 UKCA 认证。CE向UKCA的过渡期将持续至 2023年 6 月 30 日。
自 2021 年 1 月 1 日起,无论是持有 UKCA 认证或 CE 认证的医疗器械,在投放英国市场之前必须在 MHRA(Medicines and Healthcare products Regulatory Agency) 注册。
对于北爱尔兰,欧盟 MDR 和 IVDR 将分别于 2021 年 5 月 26 日和 2022 年 5 月 26 日起执行。即使在 2023 年 7 月 1 日之后,在北爱尔兰上市的医疗器械仍需持有 CE 标志,制造商需要满足欧盟法规。
1. UKCA 标志的标签和使用说明 (IFU) 要求
如果相关,务必确保标签和 IFU 已更新,体现 UKCA 标志。这方面的主要考虑因素包括:
标签必须体现 UKCA 标志,如果有授权机构参与合格评定过程,需包含该授权机构编号。
对于非英国制造商,如果使用 UKCA 标志进入英国大不列颠市场 (Great Britain),需要在标签或外包装或 IFU 上(取决于设备类型)上显示英国授权代表 (UKRP) 的详细信息。IFU 需要体现 UKCA 标志。
2023 年 7 月 1 日之前,设备标签上可同时具有 CE 和 UKCA 标志。2023 年 7 月 1 日之后,英国大不列颠市场 (Great Britain) 将继续接受双重标志。
2. 英国授权代表
如果制造商位于英国境外,必须指定一名英国授权代表 (UKRP)。对于非英国制造商所有带有 UKCA 或 CE 标志的产品,均必须符合这一要求。
UKCA合规过程中,FDASUNGO提供:
√ 英国合规负责人
√ 英国MHRA注册申报
√ UKCA的技术文件更新或者编撰
√ 英国认证机构UKCA认证评审辅导
√ 策划应对欧盟和英国市场准入最优方案
3. UKCA 标志所需的符合性声明 (DoC)
如果附上了 UKCA 标志(包括器械带有双重标志的情况),则您的 DoC 需要进行 UKCA 标志更新。DoC 应体现英国相关法律要求,包括引用的相关法律为 2002 年医疗器械法规 (SI 618),及其 2019 (SI 791) 和 2020 年 (SI 1478) 的脱欧条例修正案。
4. 英国横向立法和指定标准
作为 UKCA 的一部分,对横向法律参考(Horizontal legislation references)进行了变更。请参考图表评估您的要求。
此外,英国脱欧后,欧盟官方公报上公布的协调标准对其不再适用。因此,英国也已公布了支持本国法规的标准清单。这些标准被称为“指定标准designated standards”。
制造商若使用这些标准来证明其器械合规性,应了解英国指定标准清单的公布情况,并时刻关注有关这些清单更新的更多信息。
5. 所有医疗器械都需要在MHRA登记注册吗?包括体外诊断设备吗?
是的,所有医疗器械包括体外诊断设备都需要在英国MHRA进行登记注册并且有指定的英代(UKRP)才能在英国市场进行销售。
6. UKCA 标志是否和CE 标志一样,需要包含UK AB号?
根据UK法规的要求,如果产品需要英国批准机构做符合性评估的话,UKCA标志是需要包含批准机构号码的。
7. UKCA对于技术文档有特殊的要求吗?欧盟MDR的技术文档是否可以满足UKCA的要求?
一般来讲,MDR的要求是可以覆盖UKCA的要求,因为UKCA的要求是基于MDD。但是,企业能够熟悉不同法规对应的不同要求是很重要的。
8. I类产品进入英国市场可以自我宣称吗?需要获得UKCA证书吗?
依据UK MDR 2002法规要求,I类产品和others类IVD产品在贴上UKCA 标志和进入英国市场前需要有自我宣称的符合性声明。但I类灭菌产品和I类带测量功能产品在贴UKCA标志和进入英国市场前,需要获得UK AB的批准。
9. 爱尔兰共和国是遵守UK MDR 2002,还是EU MDR?
爱尔兰共和国是欧盟的一部分,进入爱尔兰市场需要遵守EU MDR。
澳洲TGA
TGA 对医疗器械的分类与欧盟几乎一致,且欧盟 CE 是可以被 TGA 认可的,并可以作为满足澳大利亚安全法规的重要注册资料。
为了高效监管,医疗器械被TGA按照风险级别分为5类。分别是:
Class I. (低风险)
Class IIa. (中低风险)
Class IIb. (中高风险)
Class III. (高风险)
有源植入式医疗器械(AIMD).
这5类不包含体外诊断设备(IVD),体外诊断设备有独立的分类程序。
在确认产品的医疗器械分类后,申请人(在TGA认证中被称为Sponsors)应当提交产品的技术文档给TGA,TGA将花费约200天完成审查(Class II),审查通过则签发TGA证书。
技术文档应当包括产品测试报告、技术文件以及制造商的质量体系(QMS)等。
澳大利亚TGA有条件认可欧盟CE MDR,如果申请人提供有效的CE证书,将能够用于替代部分技术文件。
同时,TGA认可MDSAP,如果制造商能够提供有效的MDSAP的审核报告,那么可以免于TGA对制造商质量体系的审核。
在TGA认证完成后,申请人将获得TGA颁发的证书。
TGA证书长期有效,但是每年需要缴纳年费。
申请人必须是澳大利亚当地公司;如果国外公司出口医疗器械到澳大利亚,则必须有当地公司作为法规联系人(也被称为当地代表)。
澳大利亚医疗用品管理局(TGA)也是TGA认证的监管机构,对于违反《医疗用品法》的行为,最高可处罚5年监禁和5000个罚款单位(约合350万RMB)。
品牌商应当谨慎保证产品合规。
加拿大CMDCAS
不同于美国,亦不同于欧洲,加拿大实行政府注册结合第三方的质量体系审查。这里所说的第三方,指经加拿大标准委员会 (SCC) 所认可的能够进行加拿大医疗器械合格评定体系审核的第三方机构。
1、不同于美国食品药品管理局(FDA)政府一手抓到底即政府的产品注册加上政府的现场审查(GMP审查),亦不同于欧洲的完全第三方公告机构(NotifiedBody)检查制度(CE认证),加拿大实行政府注册结合第三方的质量体系审查。这里所说的第三方,指经加拿大医疗器械认证认可机构(CMDCAS,CanadianMedicalDevicesConformity)认可的第三方机构,以下称CMDCAS认可机构。
2、加拿大医疗器械法规依据器械的使用风险将医疗器械分为I,II,III,IV四个分类,如I类器械为最低风险,IV类器械风险为最高。为此针对制造者提出的产品注册要求也是逐级增加,要求制造者实行的体系是愈加详尽。
审核点
对I类医疗器械(包括IVD)没有质量体系要求,并豁免产品注册许可。
加拿大医疗器械法规(Canadian Medical Device Regulations, CMDR)不要求进口商或分销商进行质量体系注册。
加拿大的现行质量体系标准为CAN/CSA-ISO 13485-03(R2013)。
II类医疗器械制造商应符合中除设计以外的要求;III和IV类器械应符合包括设计在内的所有CAN/CSA-ISO 13485-03(R2013)要求。
质量体系认证由被第三方机构签发,这些第三方机构是由Standard Council of Canada(SCC)和加拿大卫生部联合指派的医疗器械符合性评估系统内(Canadian Medical Devices Conformity Assessment System, CMDCAS)的机构。目前我国国内TUV莱茵、TUV SUD、SGS等机构具有CMDCAS资质。
与欧美医疗器械认证方式不同的是,加拿大CMDCAS认证,仅质量体系部分由加拿大认可的第三方机构进行审核,注册文件的最终审核由加拿大卫生部执行。
韩国 KFDA
韩国医疗器械法把医疗器械分为 4 类(Ⅰ、Ⅱ、Ⅲ、Ⅳ),这种分类方法与欧盟对医疗器械的分类方法非常相似。
韩国MFDS认证把医疗器械按照风险级别分为四类:(不含体外诊断设备)
Class I. 极低风险产品。如眼科显微镜、防辐射手套、手术台、听诊器等。目前有521个类别。
Class II. 低风险产品。如脉搏血氧仪、磁共振成像仪、脑电波检测仪等。目前有1017个类别。
Class III. 中等风险产品。如麻醉系统、避孕套、缝合线等。目前有318个类别。
Class IV. 高风险产品。如植入式除颤器、冠状动脉支架、可生物降解椎间盘、人工晶体等。目前有253个类别。
体外诊断设备(IVD)有独立的分类,目前也是分为4类,一共有225种。
MFDS认证必须由韩国当地公司发起申请,其申请流程和中国NMPA类似,申请人需要提交产品的测试报告、技术文件、临床报告及质量体系证明等给相应机构,在获得批准后签发MFDS证书。
其中质量体系(QMS)需要符合韩国良好生产规范(KGMP),制造商必须在经过韩国MFDS认可的机构现场审核后才能获得KGMP证书。
而测试必须在韩国MFDS认可的实验室进行,虽然目前MFDS有国外实验室认可计划,但是医疗器械测试实验室目前都位于韩国本土。
具体来说,Class I类设备适用于通知程序,Class II类设备适用于认证和批准程序,Class III和Class IV类设备仅适用于批准程序。
一般情况下,现存的Class I和Class II类设备由医疗器械信息和技术援助中心 (MDITAC)及韩国国家医疗器械安全信息研究所 (NIDS)认证;新产品以及Class III、Class IV类设备则由MFDS直接审核。
医疗器械和人体健康安全息息相关,品牌商应当严格遵守《医疗器械法》《医疗器械法执行条例》等法规。
一旦被发现有违法行为,会被按例处罚最高3000万韩元(约合30万RMB)。
日本 PMDA
医疗器械公司希望把产品投放到日本市场必须要满足日本的 PMDA,但是语言问题和复杂的认证程序还是日本医疗器械注册的一个困难点。
PMDA和MHLW
日本的药品和医疗器械管理是由卫生劳动和福利部(日本厚生省)(Ministryof Health, Labor and Welfare,MHLW)负责。
PMDA全称为Pharmaceuticals and Medical Devices Agency(独立行政法人药品和医疗器械综合机构),是MHLW管辖的独立行政法人。PMDA的业务包括审查、安全对策、健康损害救济等,对医疗器械进行技术复核和相关研究工作。
在日本,药品、医疗器械管理法律法规主要分为三类:
● 由日本议会批准通过的称法律;
● 由日本政府内阁批准通过的称政令或法令;
● 由厚生省大臣批准通过的称告示或省令。
注册流程详解
1. 确定产品分类
日本医疗器械术语集(Japanese Medical Device Nomenclature , JMDN) 编码明确了器械分类与注册登记路径。根据医疗器械的风险等级,分为一类、二类、三类和四类。
● 一类为一般医疗设备,认为即使发生不良事件,对人体的风险也极低的产品,如手术刀等。→ 须进行地方政府备案,无实质性审查。
● 二类为管制医疗设备,认为即使发生不良事件,对人体的风险也比较低的产品,如电子内窥镜、消化器官用导管等。→ 须由第三方认证机构RCB负责审查。
● 三类为高度管制医疗设备,认为在发生不良事件时,对人体的风险比较高的产品。如透析器、人工骨骼、人工呼吸器、心脏血管用球囊导管等,→ 须进行PMDA审查
● 四类为高度管制医疗设备,是对患者的侵入性高、在发生不良事件的情况下有可能直接导致生命危险的产品,如起搏器、人工心脏、支架等。→ 须进行PMDA审查
2. 任命MAH/D-MAH
所有类别器械:任命MAH或D-MAH管理日本器械上市前申请或审批。
MAH全称Marketing Authorized Holder(日本上市许可持有人),拿到某一类产品MAH执照后,才可以提出具体产品的上市申请。由于外国公司在日本没有办事处,需要任命一名在日本持有营业执照的指定上市许可持有人D-MAH (Designated Marketing Authorization),协调货物放行给外国公司的经销商以及处理投诉和警戒信息事宜。
3. 进行制造商登记
● 日本制造商向地方当局提交制造商注册 (MR) 申请。
● 外国制造商向PMDA提交外国制造商注册 (FMR) 申请。
医疗器械产品投放到日本市场必须满足日本药品和医疗器械法案(Pharmaceutical and Medical Device Act, PMD Act)。
● PMD Act要求日本本国制造商向当地机构申请其生产制造场所的注册登记,并获得制造商注册登记(Manufacturer registration, MR) 证书;
● PMD Act要求外国制造商向PMDA申请其生产制造场的注册登记,并获得外国制造商注册登记(Foreign manufacturer registration, FMR)证书。
● MR和FMR证书是提交医疗器械注册登记申请时的一项要求,提出申请前必须取得证书。
4. 质量管理体系 J-GMP
(MHLW Ordinances No.169)
● 一类器械不需要J-GMP审核。
● 二类器械由注册认证机构(RCB)进行J-GMP审核。
● 二类(除特殊控制外)、三类和四类器械,由PMDA进行QMS审核。
● 没有JMDN编码的新器械、四类器械或需要进行临床试验的器械,通常会进行现场审查。
● J-GMP是进入日本市场的医疗器械企业必须满足的、基于ISO 13485 并附加日本法律法规特殊要求的一套质量管理体系要求。
5. 提交上市申请
1. 上市前注册申请(Todokede)
一类医疗器械(必须向PMDA提交一份上市前注册申请。申请为通告性文件,PMDA不会做出任何审查意见。
2. 上市前认证(Ninsho)
有认证标准(JapaneseIndustrial Standards, JIS)的二类(及部分三类)器械必须通过上市前认证。MAH向注册认证机构(RCB)提交申请并通过认证。
3. 上市前审批(Shonin)
除了特殊控制的二类器械外的其他二类器械和三、四类器械,由MAH或DMAH向PMDA提交上市前审批申请,并获得厚生劳动省(MHLW)的批准。
6. 颁发证书
● 二类器械由RCB颁发上市前认证证书。
● 二类(除特殊控制外)、三类和四类器械由MHLW颁发上市前批准证书。
● 器械注册无有效期。
新加坡HAS
在新加坡,医疗器械产品分为A、B、C、D等4类。
A类医疗器械为低风险产品,如医用扩张器/压舌板;B类为中低风险产品,如皮下注射针/吸入式设备;C类为中高风险产品,如肺通气器/接骨板;D类为高风险产品,如心脏瓣膜/可植入除颤器。
A类医疗器械的注册申请分为4步,即提交申请、筛选、评审、主管部门作出决定。
B、C、D类产品的申请材料包括,参照东盟通用提交资料模板(CSDT)准备的英文申请,相关证书、报告和标签复印件。同时,还应提交良好的流通规范证书(GDPMDS)或医疗器械质量管理体系ISO 13485证书。申请公司应指定一名主要联系人,负责与主管部门联络与申请相关的所有问题,包括按要求补充材料。。
俄罗斯
主管当局:
俄罗斯联邦居民健康与社会发展监督部(RZN)
要求及流程:
1.确定分类,并确定命名分类代码。
2.任命一名授权代表来协调您在俄罗斯的医疗器械注册。
3. 将设器械信息和现有测试报告发送到俄罗斯的授权测试实验室,文件必须翻译成俄文。
4. 准备并向 RZN 提交进口许可申请, RZN 颁发许可证。
5. 将器械发送到俄罗斯的授权测试实验室进行质量、安全和功效测试。
6. 准备注册文档,包括技术信息、测试结果、ISO 13485 证书和现有临床数据。提交给 RZN 并支付费用。大多数文件必须翻译成俄文。证书必须经过公证和加注
7. 对于 IIa、IIb 和 III 类:RZN 进行完整性审查。如果可以接受,档案将发送到专业技术中心进行技术审查。专业知识中心审查补充临床数据要求并提出建议。RZN 审查建议并就后续步骤发表最终意见。
8. 对于 IIa、IIb 和 III 类:按照 RZN 的临床数据要求,在俄罗斯进行额外的测试或试验。
9. 对于所有器械类别:文档经过第 2 阶段审查。如果可以接受,RZN 将颁发注册证书并在 RZN 网站的注册数据库中列出产品。注册不会过期。
10.对于所有器械类别:指定俄罗斯声明人并申请符合性声明 (DoC) 认证。
巴西
主管当局:
ANVISA全称Agência Nacional de Vigilancia Sanitária,隶属巴西卫生部,负责所有医疗器械、体外诊断产品及其他健康相关产品(如药品、卫生用品、化妆品等)的上市前与上市后的管控。
要求及流程:
巴西医疗器械分类:ANVISA将医疗设备(medical equipments),用于健康的材料(materials for health use),骨科植入物(orthopedic implants)和体外诊断(in vitro diagnostics)统称为医疗器械。
巴西的医疗器械应根据其对消费者、患者、经营者或相关第三方构成的风险程度,将其划分为第I、II、III或IV类。
对于 II,III或IV类医疗器械,应提交的注册资料如:
a)相应的健康监督费的支付凭证;
b)用于识别制造商或进口商及其医疗器械的信息;
c)海外制造商或出口商授权进口商在巴西将该医疗器械商业化的授权书副本。经出口经营者授权,进口经营者应当说明生产经营者与出口经营者之间的商业关系;
d)进口医疗器械,由医疗器械生产和营销所在国主管当局颁发的注册证明或免费销售证明(或同等文件);
e)证明符合技术法规中所载的法律条文,例如管制医疗产品的ANVISA法例。
I类医疗器械注册的制造商或进口商,须向ANVISA提交上述第(a)、(b)及(e)项所列明的文件。
有源类医疗器械还需要先进行INMETRO认证,再提交ANVISA进行注册。
巴西医疗器械注册对于体系的要求:
GMP证书是ANVISA发行的证明企业符合良好生产规范的证书。出口到巴西的III,IV类医疗器械,需要符合BGMP的要求。
对于医疗器械,国际检查仅适用于制造III级和IV级风险产品的公司。外国申请人公司的巴西代表必须每两年更新一次GMP证书。但是,将进行风险分析,以决定是否需要重新检查或是否可以根据文件分析来更新证书。
阿根廷
主管当局:
国家药品、食品和医疗技术管理局(ANMAT)
要求及流程:
根据ANMAT的要求,申请批准的医疗器械需要满足以下要求:
1.指定阿根廷授权代表(AAR):ANMAT要求寻求授权的医疗器械公司必须指定一个当地代表作为整个注册过程的联络人。AAR必须获得ANMAT良好生产规范(GMP)认证,涵盖提交的设备,并持有授权许可。对于II、III、IV类器械以及IVD,AAR需要提交:
2.自由销售证书(CFS)或给外国政府的证书(CFG)
3.商业历史
4.档案资料
5.注册费支付证明
6.南方共同市场符合性声明
7.报告召回以及现场安全纠正措施的宣誓书
注:对于I类器械,AAR只需要提交付款证明、制造商信息和南方共同市场符合性声明。
8.自由销售证书(CFS)/外国政府证书(CFG):CFS或CFG必须由与ANMAT有协议的国家的认可机构(拥有医疗设备及其适用配件的信息,以及制造商的名称)提供。
9.西班牙语翻译的文件:所有提交给ANMAT的文件必须翻译成西班牙语。此外,还必须有:设备分类、使用说明(IFU)、标签、制造商的信息和技术文件。
墨西哥
主管当局:
防御卫生风险联邦委员会(COFEPRIS)
要求及流程:
1.委任一名墨西哥注册持有人(MRH)来作当地代表。
2.对证明器械获得了原产国的批准。* 满足这一要求的常用方法是使用自由销售证书(CFS,Certificate of Free Sale)或 致外国政府函(CFG,Certificate to Foreign Government)。
3.指定一名具有资质的经销商将您的医疗器械或IVD进入墨西哥。
4.对于I类低风险医疗器械:向COFEPRIS提交包括公司和医疗器械基本信息在内的申请。所有文件必须用西班牙语提交。
对于I、II和III类器械:编制详细的注册档案,包括器械/制造信息、实验室测试报告等。提供符合质量管理要求的证据(例如ISO 13485证书)和/或CE证书。
5.对于I、II和III类器械:提供特定的测试报告,具体取决于产品的功能和预期用途。
6.对于所有类别:所有器械必须遵循NOM-137-SSA1-2008的标签要求。标签和说明书必须使用西班牙语。
7.对于I、II和III类器械:MRH向COFEPRIS或第三方审查人**提交注册档案以供审查,并支付注册费用。所有文件必须用西班牙语提交。
8.对于所有类别:COFEPRIS颁发证书并在COFEPRIS网站上发布注册登记证明。注册证书的有效期为5年。有些产品可能需要进口许可证才能进入墨西哥。
其它国家医疗器械和IVD注册介绍
巴拿马、秘鲁、阿拉伯、泰国、越南、印尼、哈萨克、尼泊尔、巴基斯坦、斯里兰卡、意大利、波兰、德国、巴尔干、维也纳、埃及、苏丹、西非、沙特、迪拜、波斯、伊朗、利比亚,可以直接做CE的注册就能搞定基本所有国家。但根据产品分类不同流程有所区别。
I类的只要自我申明符合MDD指令要求就可以,III类非常繁琐,需要所有技术文件交所在国(一般是德国)主管当局审核。其他所有国家,属于欧盟以内的需要办理ISO13485及CE认证,MDI类(不灭菌、非测量)可以自我申明,不需发证;其他非欧盟以内的国家需要在CE认证的基础上办理自由销售证书(CFS)。
IVD印度注册需要准备资料有:印度使馆认证Form9(源于法规有模板),贸促会认证资料:自由销售证书FSC,质量管理体系类文件ISO13485,ISO9001,CE认证类文件EC certificate和 自我声明,标签说明书和分析报告COA。
IVD叙利亚注册需要准备资料有:SDS(SAFETY DATA SHEET),叙利亚使馆认证资料:自由销售证书FSC,质量管理体系类文件ISO13485,ISO9001,CE认证类文件EC certificate和 自我声明,营业执照,销售授权书。此外需要提交说明书。
IVD坦桑尼亚注册需要准备资料有:中国产品注册证,其他国注册证,质量管理体系类文件ISO13485,ISO9001,CE认证类文件EC certificate和 自我声明,此外需要提交COA(Certificate of analysis)。
IVD泰国注册需要准备资料有:SDS(SAFETY DATA SHEET),泰国使馆认证资料:自由销售证书FSC,质量管理体系类文件ISO13485,ISO9001,其他资料:销售授权书。此外需要提交标签,说明书。
IVD柬埔寨注册需要准备资料有:贸促会认证FSC 非认证资料:申请表 (其法规有模板),质量管理体系类文件ISO13485,ISO9001,CE认证类文件EC certificate和 自我声明,标签,说明书和销售授权函,此外每个试剂还需提交两盒样品。
IVD黑山注册需要准备资料有:贸促会认证资料:质量管理体系类文件ISO13485,ISO9001, 自由销售证书FSC,产品保险保单函。CE认证类文件EC certificate和 自我声明,非认证资料:标签,说明书和销售授权函,一套产品的GMDN & EDMS 码报告。
IVD玻利维亚注册需要准备资料有:玻利维亚使馆认证FSC 贸促会认证资料:质量管理体系类文件ISO13485,ISO9001,非认证资料CE认证类文件EC certificate和自我声明,包材和标签,COA(certificate of analysis)说明书和终产品性能评估报告,年度稳定性报告,销售授权函,一套标签及包材样品。
IVD埃塞俄比亚注册需要准备资料有:埃塞俄比亚使馆认证FSC 非认证资料:两表格Application and Consent form (其法规有模板),质量管理体系类文件ISO13485,ISO9001,CE认证类文件EC certificate和 自我声明,包材和标签,说明书和性能评估报告,稳定性报告,监测和测量程序,仪器操作手册,仪器软件确认和验证报告,仪器性能评估报告,仪器标签说明书,他国注册证,技术文件总结summary of technical documentstion (STED),基本要求检查表EP,销售授权函。
结语
不同国家或者地区对于医疗器械有不同的注册要求,国外买家会针对性地向生产商提出当地的注册要求。
在众多的注册要求中,相对而言,CE认证及FDA注册在全球范围内认可度较高,所以很多厂商会选择CE认证或者FDA注册作为打开国际市场的敲门砖,消除贸易壁垒,增加产品国际影响力,从而快速进入国际市场。