您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2023-09-19 08:31

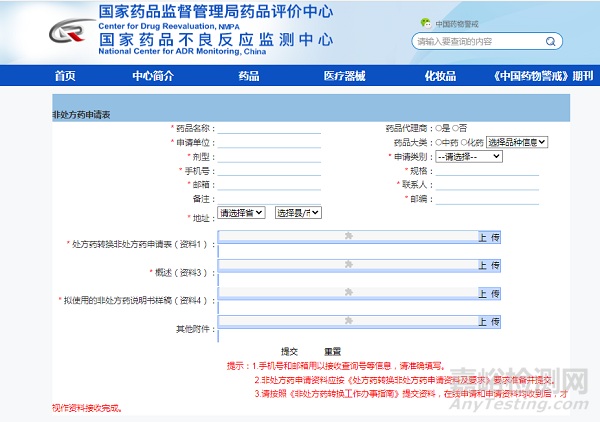
|
资料项目 |
具体资料 |
第一类 |
第二类 |
第三类 |
|
综合资料 |
1.处方药转换非处方药申请表 |
+ |
+ |
+ |
|
2.资料目录 |
+ |
+ |
+ |
|
|
3.概述 |
+ |
+ |
+ |
|
|
4.拟使用的非处方药说明书 |
+ |
+ |
+ |
|
|
5.现销售的最小销售单位样品照片 |
+ |
+ |
+ |
|
|
6.证明性文件 |
+ |
+ |
+ |
|
|
7.药品制剂及活性成份、辅料的法定质量标准 |
+ |
+ |
+ |
|
|
安全性资料 |
8.1制剂和活性成份毒理研究资料 |
△ |
△ |
△ |
|
8.2制剂和活性成份毒理文献资料 |
△ |
△ |
△ |
|
|
9.临床安全性研究资料 |
+ |
+ |
+ |
|
|
10.依赖性研究资料 |
* |
* |
* |
|
|
11.耐受性研究资料 |
- |
- |
- |
|
|
12.与其他药物和食物相互作用的情况 |
- |
+ |
+ |
|
|
13.消费者进行自我诊断、自我药疗情况下的安全性研究资料 |
- |
+ |
+ |
|
|
14.广泛使用情况下的安全性研究 |
- |
+ |
+ |
|
申报类别 |
情形描述 |
研究要求 |
|||||
|
药学研究 |
药理毒理学研究 |
临床药理学研究 |
临床研究 |
||||
|
(一)境内已有相同活性成分、适应症、剂型、规格的非处方药上市的药品 |
1.仿制境内上市的非处方药 |
1.1.所有特征一致 |
同处方药要求 |
豁免 |
豁免/BE |
豁免 |
|
|
1.2.仅改变口味、颜色、气味、清凉度、稠度、硬度、包装规格等,且变更事项不影响药品质量和疗效特征 |
同1.1.的技术要求 |
||||||
|
(二)经国家药品监督管理局确定的非处方药改变剂型或者规格,但不改变适应症、给药剂量以及给药途径的药品 |
1.在境内上市的非处方药基础上,改变剂型 |
1.1.相同释药行为的新剂型 |
同处方药要求 |
豁免 |
豁免/BE |
豁免 |
|
|
1.2.改变释药行为的新剂型 |
同处方药要求 |
豁免/必要的毒理毒代研究 |
豁免/BE或BA |
豁免/必要的桥接研究 |
|||
|
2.在境内已上市的非处方药基础上,改变规格 |
同处方药要求 |
豁免 |
豁免/BE或BA |
豁免 |
|||
|
(三)使用国家药品监督管理局确定的非处方药的活性成份组成的新的复方制剂 |
1.境内上市的非处方药活性成分组成的新复方 |
同处方药要求 |
豁免/必要的毒理毒代研究 |
必要的BE和药物相互作用研究 |
豁免/必要的桥接研究 |
||
|
(四)其他直接申报非处方药上市许可的情形 |
1.境外上市的非处方药 |
1.1.上市基础良好 |
同处方药要求 |
豁免 |
豁免 |
豁免/必要的桥接研究 |
|
|
1.2.上市基础较好,但需按照我国注册要求补充部分研究内容 |
同处方药要求 |
豁免/必要的毒理毒代研究 |
豁免/必要的临床药理学研究 |
豁免/必要的桥接研究 |
|||
|
1.3.上市基础较差,无法通过补充研究达到我国注册要求 |
不建议申报 |
||||||
|
2.仿制境外上市但境内未上市的非处方药 |
2.1.所有特征一致 |
2.1.1.被仿品境外上市基础良好 |
同处方药要求 |
豁免 |
BE |
豁免/必要的桥接研究 |
|
|
2.1.2.被仿品境外上市基础较好,但需按照我国注册要求补充部分研究内容 |
同处方药要求 |
豁免/必要的毒理毒代研究 |
BE及必要的临床药理学研究 |
豁免/必要的桥接研究 |
|||
|
2.1.3.被仿品境外上市基础较差,无法通过补充研究达到我国注册要求 |
不建议申报 |
||||||
|
2.2.在被仿品基础上,仅改变口味、颜色、气味、清凉度、稠度、硬度、包装规格等,且变更事项不影响药品质量和疗效特征 |
同2.1.1.和2.1.2.的技术要求 |
||||||
|
3. 境内已上市的非处方药,新增适应症/用药人群 |
同处方药要求 |
必要的毒理毒代研究 |
必要的临床药理学研究 |
必要的疗效验证或确证研究 |
|||
|
4. 境内已上市的非处方药,新增给药途径 |
同处方药要求 |
必要的毒理毒代研究 |
必要的临床药理学研究 |
必要的疗效验证或确证研究 |
|||
|
5.新活性成分的非处方药 |
同处方药要求 |
必要的毒理毒代研究 |
必要的临床药理学研究 |
必要的疗效验证或确证研究 |
|||
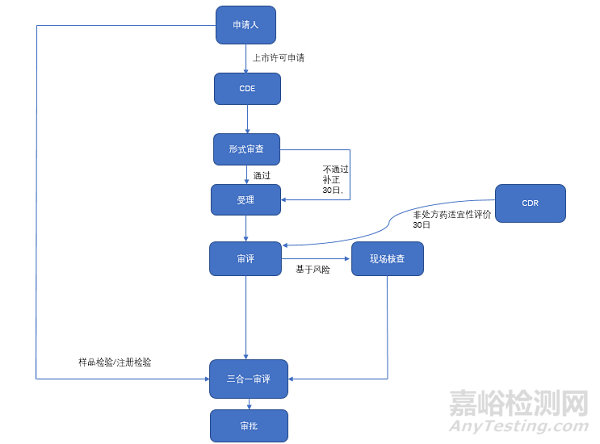

来源:注册圈


