过程分析技术(process analytical technology,PAT)是指以保证终产品质量为目的,通过对有关原料、生产中物料及工艺的关键参数和性能指标进行实时(即在工艺过程中)检测的一个集设计、分析和生产控制为一体的系统。
它最早起源于 1993 年美国分析化学家协会(AOAC International)发起的一个论坛,后在 2001 年由美国食品药品监督管理局 (Food and Drug Administration,FDA)药品评价与研究中心主任 Janet Woodcock 博士总结提出了 PAT 的倡 议。
2004 年 FDA 正式发表了关于 PAT 的工业指导原则——Guidance for industry PAT——A framework for innovative pharmaceutical development,manufacturing, and quality assurance,拉开了 PAT 在制药领域应用的序幕。
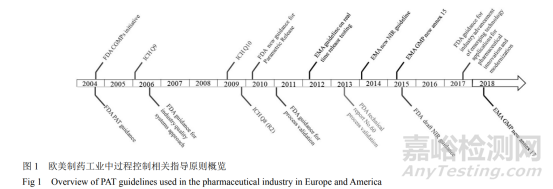
该技术不仅可以缩短工时、减少误差,还可以使监管更加有据可依,因此越来越多的欧美制药企业开始引入 PAT 以实现药品生产过程的全程监控,从而保证产品的质量。同时,为了推广和规范 PAT 技术,这些国家和地区的药品监管部门,也先后出台了许多相关的标准和指导原则(见图 1)。
本文将目前欧美等国家与 PAT 概念、方法、应用和监管相关的主要标准和指导原则加以总结和分析,以帮助国内制药领域更好地理解和应用这项技术。
美国的相关举措
FDA 作为全球制药领域的风向标,成为最早发起在制药领域实施 PAT 的监管机构。
FDA 在制药领域提出 PAT 的倡议,并非心血来潮,而是各方利益冲突的撞击与发展。
随着制药行业的迅速发展,美国制药工业界认为 FDA 过于严格,使企业在生产过程中缺乏灵 活性;而 FDA 则面临人员短缺但药物申请的文件审查、 现场认证以及突发事件处理等越来越繁重的负担,加之公众对于高质量药品的期待,为了协调三方的矛盾, FDA 在 2002 年发起了一个新的倡议——Pharmceutical CGMPs for the 21st century——a risk-based approach,并于 2004 年形成最终报告,告知制药企业和公众,FDA 将 建立更加科学的基于风险管理的高效监管新策略,鼓励制药企业使用新的技术:
如 PAT 来促进药品的连续生产以及质量的提高和创新,并成立 PAT 工作组加强监管 部门与制药企业相关方面的沟通和合作,推进 PAT 的 实施。该报告还首次提出了药品生产领域一个全新的理 念——质量源于设计(Quality by Design,QbD),该理念为 PAT 等创新技术的实施提供了监管和技术架构。
随后 FDA 在 2004 年发表了 PAT 工业指导原则 Guidance for industry PAT——A framework for innovative pharmaceutical development,manufacturing, and quality assurance。
该指导原则首先介绍了在制药领域推行 PAT 的背景、目的以及 PAT 的定义,然后系统地介绍了制药企业实施 PAT 需要获得哪些方面的知识,FDA 为了推行 PAT 做了哪些努力(包括成立 PAT 专家组,PAT 培训等等),以及制药企业应如何与 FDA 合作、沟通、提交资料以获得实施 PAT 的批准。
从该指导原则可知,PAT 不是一项单纯的技术而是一 个技术领域,制药企业实施 PAT 不仅需要对生产过程有深刻的理解,还需要了解和掌握与 PAT 相关的知识 和工具。
PAT 工具主要包括以下四类:用于设计、数据采集和过程分析的多变量分析工具;现代过程分析仪器和过程分析化学工具;过程控制工具以及持续的改进和知识管理工具。另外在实施 PAT 时还应掌握风险管理方法、系统集成方法和实时放行方法。
其实早在 FDA 提出 PAT 实时放行方法以前,在欧美等无菌药品生产领域已经用实际行动在践行这一理念——参数放行(Parametric release)。
所谓参数放行就是一种基于对业已通过验证的灭菌程序的有效质量控制、监测和系统文件管理来替代依赖于最终产品检验的无菌放行程序。
由于无菌检验方法的统计学限制,最终产品的无菌检验只能提供一个发现无菌保证系统错误的机会,无菌药品的无菌特性并非依赖于最终成品的检验,而是取决于对药品生产全过程严格的质量管理。
1987 年,FDA 正式颁布了参数放行的工业指南,参数放行正式进入药品生产企业的生产质量管理规范(Good Manufacturing Practices,GMP)。
最新版本为 2010 年 2 月颁布的终端灭菌产品实施参数放行的相关申报资料要求 Guidance for industry submission of documentation in applications for parametric release of human and veterinary drug products terminally sterilized by moist heat processes,经过多年的不断完善,发达国家的药品监管部门对于无菌药品已经普遍接受了参数放行的理念并付诸实践,分别制定了相应的指导原则。
美国、加拿大、澳大利亚等国家以及欧盟对参数放行的批准及日常监管内容和形式基本一致,都是在现行 GMP 管理的基础上颁布参数放行指南和申报办法,由企业自愿申请,药品监管部门进行严格的资料审核和现场检查后,决定是否批准企业的申请,已获批准的药品如发生重要因素的变更需重新申请。
为了推行更加科学的监管理念、鼓励制药企业不断创新和发展,FDA 也一直在积极地寻求与国内外研究、生产和监管领域的合作。
当 FDA 提出 QbD 理念后,很快被人用药品注册技术规定国际协调会(The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH) 所 采 纳, 与 FDA 联合发布了 ICH Q8(R2)(Guidance for industry Q8(R2)Pharmaceutical development)、ICH Q9(Guidance for industry Q9 Quality risk management) 和 ICH Q10(Pharmaceutical quality system),这 3 个指导原则具体表达了过程和产品开发、 技术转移和商品生产中 QbD 的理念。
所谓 QbD 的理念就是从产品和生产工艺的研发阶段开始进行合理的实验设计,研究产品质量属性与原料质量属性和生产过程参数之间的关系,建立能满足产品性能且工艺稳定的设计空间,从而在生产阶段建立一种可以在一定范围内调节偏差来保证产品质量稳定性的措施,并在商业化大生产开始后对工艺进行连续的改进。
由此可见,尽管 QbD 不等同于 PAT,但 PAT 是实现 QbD 优点的实质要素和关键驱动因素。其中 ICH Q8(R2)系统地介绍了 QbD 的基本原则以及在药品及其生产工艺的研发过程中如何应用科学策略和质量风险管理策略获取对产品和生产工艺的全面理解,从而细化了 PAT 实施前需要对生产过程深刻理解的具体范围和要求。
同时 ICH Q8(R2)在 PAT“实时放行”方法后加上“检测”二字, 即实时放行检测,将关注点转移到测量方法上,相比成品检测,实时放行检测在生产过程中进行质量监测, 将质量控制从生产过程的终点向上游转移,明确了 PAT 的优势。
ICH Q9 重点介绍了质量风险管理的原则和常用分析工具,它是实施 PAT 所需的理论基础之一, 但其内容适用范围不只局限于 PAT。
尽管 PAT 等新兴的技术前景可观,但制药企业仍会对采用新技术犹豫不决,主要原因是担心具体监管措施不明朗而导致新技术的应用延迟。为此,FDA 也在 PAT 实施细则方面进行了很多努力。
2006 年,FDA 发布了 制药企业 CGMP 规范的质量体系工业指南——Quality systems approach to pharmaceutical CGMP regulations, 旨在帮助制药企业建立以科学和风险管理为基础的质量体系来满足现行动态生产质量管理规范(Current Good Manufacturing Practices,CGMP)的要求。
该指南建议制药企业建立包含管理职责、资源、生产运作和评估活动 4 个要素在内的全面质量体系,并对每个要素的组成、职责进行了详细的建议。
FDA 认为拥有完善质量体系和恰当生产工艺的制药企业能够进行多种类型的改进,能够为“设计的质量”、持续的改进以及药品生产过程中的风险管理提供必要的框架。
在生产运作要素内,FDA 认为尽管对于某些生产过程可以用时间来界定终点,但企业应当有能力利用在线参数建立生产控制,这些参数是以实时检测和监测设备测定的预期工艺终点为基础的。
该指导原则为 PAT 在现行生产控制中的应用找到了切入点并明确了其在质量体系中所处的位置。
2011 年 FDA 整合了新的监管理念,发布了改版的工艺验证指南——Guidance for industry process validation:general principles and practices,工艺验证是法律规定制药企业必不可少的生产活动之一,该指南对工艺验证的概念和范畴进行了广度和深度的拓展,将 FDA 对于工艺验证的要求,从原先对 3 个生产规模批次样品生产过程的验证,变成了涉及整个产品生命周期和生产中发生的一系列活动的验证。
其中 FDA 建议可以使用 PAT 的工艺控制策略,由于 PAT 工艺被设计用来实时检测一种在加工材料的多种属性,因此需要使用不同的工艺性能确认方法,重点关注待检测属性的检测系统和控制环。
工艺性能确认实际上就是为证明工艺可重现且可以始终如一的生产出优质产品寻找科学依据的过程,但该指导原则对于 FDA 对工艺验证不同阶段文件的要求和期望并未给出具体的描述。
2013 年美国注射剂协会(Parenteral Drug Association,PDA) 发布了题为 Technical report No.60 process validation:A lifecycle approach 的第 60 号技术报告,对 FDA 关于产品生命周期内工艺验证的全过程进行了具体的诠释。
该技术报告的核心理念是“工艺验证不是一个一次性的活动,而是一个贯穿于整个产品生命周期,连接工艺开发与商业化生产,并在日常商业化生产中维持的活动”。它为 FDA 2011 年版工艺验证指南在制药领域实施画出了具体的线路图。
该技术报告在第六部分——实施工艺验证的系统和技术专门用了一节讲述 PAT 系统的选择以及 PAT 系统设计阶段工艺验证的考虑要点等制药企业关注的具体问题。
2015 年,FDA 又发布了工业界开发和申报近红外分析方法指导原则草案——Development and submission of near infrared analytical procedures,Guidance for industry,Draft guidance——近红外分析方法是 PAT 最常用的分析技术之一,该指导原则给出了制药行业在新药申请、简化新药申请以及药物主文件中采用近红外分析方法的开发、验证和变更需要考虑的问题以及需提交的信息。
同年,FDA 药品审评与研究中心下属的药品质量办公室还启动了新兴技术项目(Emerging Technology Program),期望制药企业从早期就能参与 该项目,通过与 FDA 的新兴技术团队召开更多会议、 进行更多沟通探讨,共同研究新生产技术中的相关问题,从而在提交注册资料之前就能发现并解决这些问题。
FDA 更是于 2017 年发布了新兴技术应用的先进性使得药品生产基础现代化的工业指南——Guidance for industry advancement of emerging technology applications for pharmaceutical innovation and modernization,对制药企业如何参与 FDA 的新兴技术项目来推动包括 PAT 等新兴生产技术的应用进行了详细解读。
美国药典(The United States Pharmacopeia,USP) 委员会是为在美国境内生产和销售的处方及非处方药物、食物补充剂和其他保健产品制订质量标准的法定机构,为协助 FDA 推进 PAT 技术,美国药典委员会也进行了大量的工作。
其中两项主要的工作是修订现有 USP 标准中有关近红外的章节(< 1119 > Near-Infrared Spectroscopy),强调了其作为 PAT 应用的要求;增 加了 PAT 另一项常用技术拉曼光谱的章节(< 1120 > Raman Spectroscopy)。
与近红外光谱相似,拉曼光谱具有快速、样品预处理简单等优势,同时它受水分干扰 小,无需复杂建模使其逐渐成为 PAT 的新宠。最新 版 USP41 通则 1120 对拉曼光谱的应用范围、影响因素、仪器以及性能确认等进行了详细的说明和要求。
欧盟的相关举措
欧盟作为制药领域的又一发达地区,其制定的标准和指导原则也经常成为大家观摩的范本。
2014 年欧洲药品管理局(European Medicines Agency,EMA) 发布正式版 Guideline on process validation for finished products information and data to be provided in regulatory submissions,该指南在 2016 年 又 做 了 小 的 修 改。
与 FDA 的工艺验证指南将工艺验证划分为 3 个阶段(工艺设计、工艺确认和持续工艺验证)不同,EMA 的这项验证指南仅适用于制剂,且仅对应于 FDA 定义的第二阶段——工艺确认。
在 EMA 的工艺验证指南中引入了连续工艺确认(Continuous process verification,CPV), 这一概念来源于 ICH Q8,当一种产品是通过 QbD 方法 开发并已经科学地建立了能高度保证产品质量的工艺控制时,可以采用 CPV 替代传统工艺验证。
CPV 是通过建立一个工艺确证体系,对物料属性、关键质量属性和关键工艺参数建立了科学的控制策略,从而保证终产品的质量。
指南中明确指出:控制策略应是经过常规评估的,可以选用 PAT 和多变量统计过程控制(Multivariate statisitical process control,MSPC)作为控制策略的工具。
对于不同的生产步骤还可以选择采用传统工艺验证和连续工艺验证混合的方式,在申报文件中应提供使用混合方法的论证并指出在哪些生产步骤采用了混合方法。
随后的 2015 年 3 月欧盟委员会发布了最新改版的欧盟 GMP 附录 15 确认与验证 Volume 4 EU Guidelines for good manufacturing practice for medicinal products for human and veterinary use Annex 15:qualification and validation,将工艺验证拓展到产品的全生命周期,对企业如何进行确认与验证进行了具体的表述,其中对于企业采用传统工艺验证方法、连续工艺确认方法或混合方法的表述与 EMA 制剂工艺验证指南基本一致。
欧盟也是较早对无菌产品使用参数放行的地区之一, 2012 年 EMA 颁布了新版的实时放行检测指南 Guideline on real time release testing(formerly guideline on parametric release)以取代 2001 年颁布的参数放行指南。
该指南指出实时放行不再只适用于无菌药物的灭菌过程,而是适用于化学药和生物制剂原料、中间体和制剂的各个生产过程。该指南还给出了实施实时放行的法规基础、应用 实例以及申请批准需要的文件材料。
2018 年 4 月欧盟委员会发布了最新改版的欧盟 GMP 附录 17 实时放行检测和参数放行 Volume 4 EU Guidelines for good manufacturing practice for medicinal products for human and veterinary use annex 17:real time release testing and parametric release,同 样在原有参数放行的基础上增加了实时放行的内容,并对企业如何实施实时放行给出了具体的要求。
关于近红外光谱技术在制药企业应用的探讨在欧洲起步较早,英国、荷兰、法国等研究机构都有过相关的应用指导原则出台,其中 EMA 制定的制药工业近红外光谱技术应用、申报和变更资料要求指南 Guideline on the use of near infrared spectroscopy by the pharmaceutical industry and the data requirements for new submissions and variations 在 2003 年发布后又根据法规与技术的变化进行了更新,于 2014 年发布新版。
它在建立、验证、变更近红外方法方面为药品生产企业提供了详细的指导,并规定了在新药申报或方法变更时,使用近红外方法需要提交的数据,该指导原则在内容设置上较 FDA 的近红外指导原则更加具体,此次的更新版本还特别在方法通用要求、定性 / 定量模型分类要求等方面针对 PAT 的应用进行了说明。
欧洲药典(The European Pharmacopoeia,EP) 是欧洲药品质量检测的唯一指导文献,它由欧洲药品质量管理局(European Directorate for the Quality Control of Medicines,EDQM)出版发行。EP 是最早收录近红外光谱技术(2.2.40. Near-infrared spectroscopy)的药典。
2018 年 1 月,EDQM 又在欧洲药典论坛上发布了 PAT 草案(5.25. Process analytical technology),计划将其收入 EP。该草案对 PAT 的定义、测量方式以及数据的分析方法等进行了简单介绍。为了推动和支持 PAT 技 术的应用,EDQM 还新增或修订了 EP 中 PAT 相关的 9 个章节(见表 1),主要涉及 PAT 的常用技术与数据分析方法。
草案也分别就这 9 个章节的修订内容进行了介绍。
如1. 通则的修订:1.1 部分说明一个物料可基于产品设计结合其控制策略和从生产工艺验证研究中所生成的数据证明其具备药典质量。同时还说明加强质量控制的方法可利用 PAT 和 / 或实时放行检测策略(包括参数放行)替代仅检测终产品的方法。
再如章节 2.2.40 近红外光谱部分:增加近红外近线、在线和原位测试的表述, 同时在样品准备 / 代表性部分增加了清除样品以及在不同测量模式中使用光纤探头系统。如果转移光纤探头用于背景光谱测量困难,可以使用内置参照用于过程分析,同时还根据仪器的测量模式和使用位点提供了详细的仪器性能控制方法。另外在定性分析部分除常规的鉴别分析外,还介绍了限度分析(如用于控制干燥终点) 和趋势分析(如混合均匀性监测)。
总结与展望
由上述欧美制药领域 PAT 相关的标准和指导原则可见,PAT 技术自 2002 年 FDA 在制药领域提出至今,已经逐步从最初的设想向实用化方向迈进。
我国在该领域的应用起步较晚,且大都集中在中药的过程控制领域。尽管 2016 年发布的《医药工业发展规划指南》和 《智能制造工程实施指南(2016 - 2020)》均将 PAT 作为未来医药领域发展主要任务之一,但有关 PAT 的具体应用指南和监管措施鲜有发布。
现行《中国药典》(2015 年版)虽已经收录近红外光谱和拉曼光谱技术,但在内容设置上主要还是针对成品的定性和定量分析,未涉及药品生产过程的监控环节。
由于多种因素的制约,无论是 PAT 技术在制药领域的应用还是监管,我国与欧美之间仍然存在很大的差距。相信,随着 QbD 理念的不断深入,制药企业终将理解以风险管理为基础的 PAT 技术是了解生产工艺、控制产品关键质量属性的最有效手段。
可以预见,随着国内制药领域 PAT 的深入开展, 药品生产企业和监管部门对 PAT 技术的逐步采纳和认可,其在药物分析领域的标准和原则也将不断完善和成熟。
来源:《中南药学》2019 年 9 月 第 17 卷 第 9 期
原标题:《欧美制药工业中过程控制主要标准和指导原则简介》
作者:冯艳春,肖亭,胡昌勤(中国食品药品检定研究院)