您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-04-20 09:40
各位小伙伴在做药品微生物限度检查的实验过程中是否遇到了很多棘手的问题?今天就带大家一起来分析一下实验过程中常见的问题。
1、微生物限度检查的基本内容
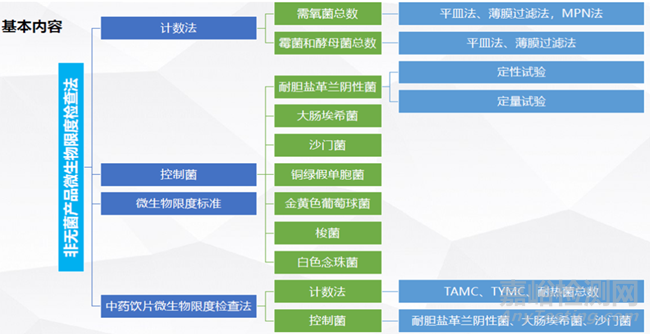
2、是否所有非无菌产品均应进行微生物限度检查?
对于《中国药典》制剂通则项下有微生物限度要求的制剂,微生物限度为必检项目;对于只有原则性要求的制剂(如部分化学药品的丸剂、口服片剂、胶囊剂、颗粒剂),应对其被微生物污染的风险进行评估。
制定药品的微生物限度标准时,除了依据“非无菌药品微生物限度标准(通则1107)”外,还应综合考虑原辅料来源、性质、生产工艺条件、给药途径及微生物污染对患者的潜在危险等因素,提出合理安全的微生物限度标准,如特殊品种以最小包装单位规定限度标准。必要时,某些药品为保证其疗效、稳定性及避免对患者的潜在危害性,应制定更严格的微生物限度标准,并在品种项下规定。
微生物限度
以动物、植物、矿物来源的非单体成分制成的片剂,生物制品片剂,以及黏膜或皮肤炎症或腔道等局部用片剂(如口腔贴片、外用可溶片、阴道片、阴道泡腾片等),照非无菌产品微生物限度检查:微生物计数法(通则1105)和控制菌检查法(通则1106)及非无菌药品微生物限度标准(通则1107)检查,应符合规定。规定检查杂菌的生物制品片剂,可不进行微生物限度检查。
以动物、植物、矿物质来源的非单体成分制成的胶囊剂,生物制品胶囊剂,照非无菌产品微生物限度检查:微生物计数法(通则1105)和控制菌检查法(通则1106)及非无菌药品微生物限度标准(通则1107)检查,应符合规定。规定检查杂菌的生物制品胶囊剂,可不进行微生物限度检查。
3、微生物限度检查培养基适用性合格,能否有效保证实验结果准确?
a、培养基适用性是关键控制点。
b、考虑其他影响因素,如对特定菌检出。
c、考虑储存条件,有效期,水分等影响。
倾注平皿时培养基通常为15-20ml,应建立质量管理体系进行较全面的质量控制,确保结果准确。
4、微生物限度检查方法适用性试验中,如何去除药品的抑菌性?

5、基质影响样品的溶解,能否直接进行适用性试验?
样品可以不溶解,但应该分散均匀,可使用表面活性剂使制备的样品分散均匀。
6、样品试验时,本底生物负载水平较高影响实验,如何处理?
a、更换样品重新进行适用性试验。
b、供试液制备时加热处理降低负载。
c、采取其他方式降低生物负载水平。
生物负载水平较低时无需处理,回收试验时减去即可,生物负载水平较高时应应该适当处理。
7、在日常微生物限度检查中,应该做几个稀释级?
a、考虑微生物限度检查的历史数据。
b、考虑其是否为新产品或原辅料。
c、考虑产品、方法、供应商等是否变更。
估计负载水平,选择适宜稀释级;无法估计负载水平时,一般选择3个连续稀释级进行试验。

来源:Internet


