您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-06-06 12:02
辅助生殖用显微操作管是指体外受精(IVF)显微操作中,用于注射、持卵、剥离、活检、辅助孵化和取精用的微细管状和针状工具。
根据《医疗器械分类目录》,辅助生殖用显微操作管属于子目录18妇产科、辅助生殖和避孕器械项下的产品,其分类编码为18-07-03,管理类别为II类。
一、辅助生殖用显微操作管的结构组成
显微操作管由针柄、针颈、针尖组成。通常由硼硅酸盐玻璃或聚碳酸酯等高分子材料制成。产品形式主要有单精子注射管、持卵管、活检管、剥卵管、打孔管等。
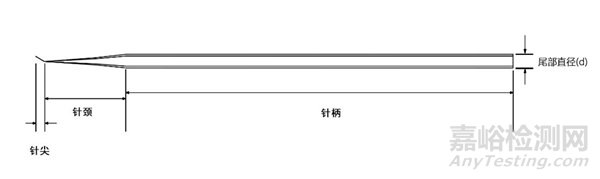

图1单精子注射管结构示意图
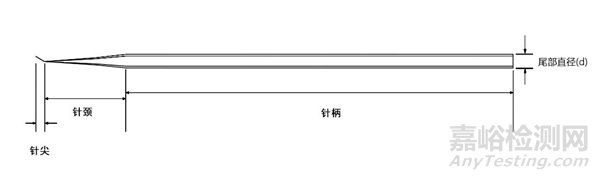
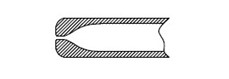
图2 持卵管结构示意图

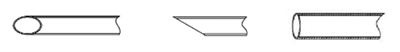
图3 活检管结构示意图


图4 剥卵管结构示意图

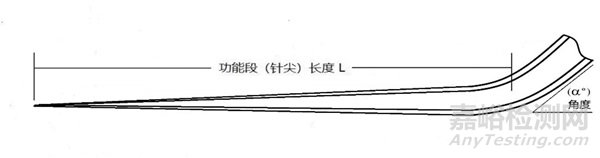
图5 打孔管结构示意图
二、辅助生殖用显微操作管的主要风险
辅助生殖用显微操作管的风险包括但不限于以下内容:
1、原材料的危害
接触部位的材料(包括与产品直接接触的初包装)或材料来源变化;材料的生物相容性对配子、合子或胚胎的影响。
2、产品设计可能产生的危害
显微操作管的尖端设计、尺寸规格、边缘光滑程度及断面的处理等对注射效率和合子存活率的影响,以及玻璃材质产品因尖端设计不当所导致操作中易崩折产生微屑、尖刺等危害。
3、生产加工过程可能产生的危害
在切割及磨针过程中残留的玻璃碎片;加工时产生气泡;污染;微粒残留;添加剂、加工助剂、辅剂的残留;工艺用水;生产环境洁净度;热原;细菌内毒素。
4、产品使用风险因素
规格选择与使用不当,或未按照说明书中操作方法操作,造成配子、合子或胚胎损伤。
5、产品包装可能产生的危害
如包装破损、标识不清;包装对灭菌方式不适宜;运输过程中无法为产品起到充足的保护等。
6、灭菌过程可能产生危害
如灭菌方式对产品不适宜、灭菌不完全、灭菌过程残留的有害物质(如环氧乙烷)等。
三、辅助生殖用显微操作管性能研究实验要求
1、物理和化学性能研究
1.1明确所有性能指标及检验方法的确定依据及采用的原因及理论基础。
1.2应明确细菌内毒素限量及其设定依据,明确环氧乙烷及其他杂质的限量及其设定依据。
对产品具有的特定性能还需开展相应的研究,如:
产品内外壁及针尖边缘光滑程度是否会损伤配子、合子或胚胎;针尖弯角、口径是否满足细胞固定或注射的要求,不会导致细胞固定不牢固而在操作过程中发生旋转或变形;针尖与针颈的距离是否适宜,不会干扰注射操作过程的稳定性。
对于其他特殊性能或需与其他器械联合使用以实现预期用途的产品,需开展设计验证。
2、生物学特性研究
成品中预期与患者、配子、合子或胚胎直接或间接接触的部分,均需要进行生物相容性评价。
生物相容性评价研究需明确:
2.1生物相容性评价的依据和方法。
2.2产品所用材料及与配子、合子或胚胎接触的性质。
接触性质需提供产品预期与配子、合子或胚胎接触的情况,包括接触部位、接触方式、接触时间(提供预期的最长接触时间)。
2.3实施或豁免生物学试验的理由。
2.4对于现有数据或试验结果的评价。
参考GB/T 16886系列标准、YY/T 1434、YY/T 1688、YY/T 1535,需考虑进行的生物相容性评价/试验项目包括热原、细胞毒性、致敏、刺激、遗传毒性、体外鼠胚试验、囊胚细胞染色和计数试验、人精子存活试验(如适用)。
开发人应选择无致癌、致畸及致突变性,无生殖毒性和遗传毒性的原材料或成分。若申报产品的材料或成分具有上述风险或相关风险不明,开发人应开展产品致癌性、致突变性及生殖毒性的生物相容性评价。
3、灭菌研究
明确申报产品灭菌方法的选择依据、灭菌工艺和无菌保证水平(应达到1×10-6),并开展灭菌确认。灭菌过程的选择应考虑以下因素:产品与灭菌过程的适应性,包装材料与灭菌过程的适应性等。
若采用湿热灭菌,应符合GB 18278.1的要求;若采用辐射灭菌,应符合GB 18280.1、GB 18280.2的要求;若产品采用环氧乙烷灭菌等其他灭菌工艺,应符合GB/T 19974、GB 18279.1、GB/T 18279.2的要求。
若灭菌使用的方法容易出现残留,如环氧乙烷灭菌,应当明确残留物限量要求及采取的处理方法,并开展研究。
5、稳定性研究
5.1货架有效期
包括货架有效期和包装研究,证明在货架有效期内,在规定的运输贮存条件下,产品可保持性能功能满足使用要求,并保持无菌状态。
开发人需开展产品有效期研究,可采用加速老化或实时老化方式展开研究,具体方法和要求可参考《无源植入性医疗器械稳定性研究指导原则(2022年修订版)》和YY/T 0681.1《无菌医疗器械包装试验方法第1部分:加速老化试验指南》等标准。实时老化研究应从产品定型后开始进行。
开发人可依据相关标准,如GB/T 19633《最终灭菌医疗器械包装》、YY/T 0681《无菌医疗器械包装试验方法》系列标准、YY/T 0698《最终灭菌医疗器械包装材料》系列标准进行产品包装验证。
5.2运输稳定性
应当开展运输稳定性和包装研究,证明在规定的运输条件下,运输过程中的环境条件不会对医疗器械的特性和性能,包括完整性和清洁度,造成不利影响。
6、其他
该产品列入《免于临床评价医疗器械目录》,开发人应当按照《列入免于临床评价医疗器械目录产品对比说明技术指导原则》的要求,开展相关研究,从基本原理、结构组成、性能、安全性、适用范围等方面,证明产品的安全有效性。申报产品与对比产品存在差异的,还应开展差异部分对安全有效性影响的分析研究。

来源:嘉峪检测网


