您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-08-30 08:33
摘 要: 建立了一种柱前衍生化-高效液相色谱法测定透明质酸钠凝胶敷料中透明质酸钠的含量,可有效实现复杂组成透明质酸钠凝胶产品的质量控制。采用强酸将透明质酸钠凝胶敷料中的透明质酸钠水解成氨基葡萄糖和葡萄糖醛酸,利用1-苯基-3-甲基-5-吡唑啉酮与单糖还原性末端反应的原理,将氨基葡萄糖衍生化后,采用高效液相色谱法进行检测。采用Kromasil C18柱分离,以乙腈-0.025 mol/L乙酸铵溶液(pH 4.5)作为流动相进行梯度洗脱,流速为1.0 mL/min,柱温为40 ℃。透明质酸钠的质量浓度在20~60 μg/mL范围内与色谱峰面积线性关系良好,相关系数为0.999 6,平均加标回收率为98.47%,测定结果的相对标准偏差为1.1%(n=9)。该方法受产品其他处方组成干扰影响小、准确度高、精密度好,可实现快速准确地测定复杂组成透明质酸钠凝胶敷料中的透明质酸钠的含量。
关键词: 透明质酸钠; PMP衍生化; 含量; 高效液相色谱法
透明质酸是由D-葡萄糖醛酸和N-乙酰氨基葡萄糖胺通过β-1,3糖苷键连接成双糖结构单元,每个双糖单元通过β-1,4糖苷键相连形成的链状粘多糖。透明质酸多以钠盐形式广泛应用于食品、化妆品、药品、医疗器械等领域[1-3]。2022年11月国家药监局发布了修订《关于医用透明质酸钠产品管理类别的公告》,明确了不同预期用途和工作原理的透明质酸钠的边缘产品、药械组合产品的管理属性和管理类别,加强透明质酸钠的产品监督管理。目前含有透明质酸钠组分产品种类繁多[4-5],制剂组分复杂,因此有必要建立一种能准确测定复杂组分产品中透明质酸钠含量的方法,为透明质酸钠产品质量控制提供方法支撑。
透明质酸钠检测方法主要包括硫酸-咔唑法[6-7]、酶解法[8]、凝胶色谱法[9]、近红外光谱法[10]。硫酸-咔唑法易受透明质酸钠产品组分中能与硫酸咔唑反应显色物质的干扰,如葡萄糖、海藻糖等;凝胶色谱法不适用于含有不同相对分子质量的透明质酸钠产品、且分析成本较高;酶解法将透明质酸钠水解成不饱和双糖,采用高效液相色谱法进行分离测定,酶解法和近红外光谱法主要适用于透明质酸钠原料的检测,对于复杂制剂处方成分透明质酸钠产品目前暂无文献报道。
透明质酸钠在强酸条件下水解可生成带有还原性末端的氨基葡萄糖和葡萄糖醛酸[11-12]。在碱性条件下,氨基葡萄糖和葡萄糖醛酸均可以与1-苯基-3-甲基-5-吡唑啉酮(PMP)发生结合反应,1个单糖分子的还原端可与2个分子的PMP形成稳定的衍生物[13-15],该衍生物改变了单糖分子的极性、具有较强紫外吸收,可采用液相色谱分析[16-18]。笔者建立了一种高效液相色谱分析方法,用于分离PMP-氨基葡萄糖衍生物和PMP-葡萄糖醛酸衍生物,并以PMP-氨基葡萄糖衍生物作为透明质酸钠的定量峰,用于透明质酸钠凝胶敷料中透明质酸钠的含量测定,该方法可以排除制剂处方中多种辅料的干扰,包括葡萄糖醛酸,海藻糖以及其他多种辅料成分,专属性强,为复杂组分透明质酸钠产品中的透明质酸钠含量测定提供了方法参考。
1、 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:LC-2030C 3D型,附带DAD检测器、LC-Solution工作站,日本岛津企业管理(中国)有限公司。
单级四级杆高效液相色谱质谱联用仪:LC-MS 2020型,附带LC-20AD高效液相色谱仪、DAD检测器、质谱检测器(DUIS复合离子源)、LC-Solution工作站,日本岛津企业管理(中国)有限公司。
电子分析天平:QUINTIX35-1CN型,感量为0.01 mg,赛多利斯科学仪器(北京)有限公司。
电热恒温鼓风干燥箱:DHG-9070A型,上海齐欣科学仪器有限公司。
pH(酸度)计:FiveEasyPlus型,梅特勒-托利多科技(中国)有限公司。
恒温水浴锅:HH-6型,常州国华电器有限公司。
超纯水机:PURELAB flex2型,威立雅水处理技术(上海)有限公司。
离心机:DMO412型,赛洛捷克(美国)有限公司。
微型涡旋仪:WH-2型,上海沪西分析仪器厂。
盐酸、氢氧化钠:分析纯,南京化学试剂股份有限公司。
1-苯基-3-甲基-5-吡唑啉酮(PMP)、D-氨基葡萄糖盐酸盐、D-葡萄糖醛酸:优级纯,上海阿拉丁生化科技股份有限公司。
甲醇、乙腈、二氯甲烷:色谱纯,霍尼韦尔(中国)有限公司。
透明质酸钠对照品:纯度(质量分数)为97.5%,山东众山生物科技有限公司。
透明质酸钠凝胶敷料样品、空白凝胶样品:江苏海智生物医药有限公司。
实验用水为去离子水,由威力雅超纯水水仪制得。
1.2 仪器工作条件
1.2.1 色谱仪
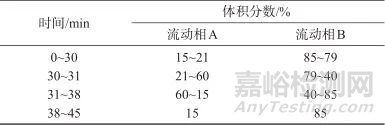
色谱柱:Kromasil 100-5型C18柱[4.6 mm×250 mm,5 μm;诺力昂新材料(苏州)有限公司];柱温:40 ℃;进样体积:10 μL;检测波长:250 nm;流动相:A相为乙腈,B相为0.025 mol/L乙酸铵溶液(用乙酸调pH至4.5),流量为1.0 mL/min,梯度洗脱,洗脱程序见表1。
表1 梯度淋洗程序
Tab. 1 Gradient washing program
1.2.2 质谱仪
离子源:加热型DUIS复合源,可同时进行电喷雾电离(ESI)和大气压化学电离(APCI);电离模式:正负离子模式同时进行;检测模式:多反应监测(MRM)模式;雾化气:高纯氮气;雾化气流量:1.5 L/min;接口温度:350 ℃;加热模块温度:400 ℃;检测器电压:1.5 kV;紫外检测器-质谱检测器柱后串联分流比:1∶1;质荷比(m/z)扫描范围:100~800;扫描速率:715 。
1.3 溶液配制
透明质酸钠对照品储备溶液:精密称取透明质酸钠对照品80 mg于20 mL容量瓶中,加入适量纯水,于70 ℃水浴中充分溶胀溶解后,加水稀释至标线,摇匀,配制成质量浓度为4 mg/mL的透明质酸钠对照品储备溶液。
透明质酸钠对照品溶液:精密量取质量浓度为4 mg/mL透明质酸钠对照品储备溶液0.5 mL于20 ml顶空瓶中,加入5 mol/L盐酸溶液5 mL,压盖,涡旋混匀后置于121 ℃烘箱中,加热水解1.5 h,取出冷却至室温后,用纯水完全转移至50mL容量瓶中,加入5 mol/L氢氧化钠溶液5 mL中和,再加水稀释定容至标线,摇匀,配制成质量浓度为40 μg/mL的透明质酸钠对照品溶液。
D-氨基葡萄糖盐酸盐对照品溶液:精密称取D-氨基葡萄糖盐酸盐10 mg于250 mL容量瓶中,加水溶解稀释,定容至标线,摇匀,配制成质量浓度为40 μg/mL的D-氨基葡萄糖盐酸盐对照品储备溶液。
D-葡萄糖醛酸对照品溶液:精密称取D-葡萄糖醛酸10 mg于250 mL容量瓶中,加水溶解稀释,定容至标线,摇匀, 配制成质量浓度为40 μg/mL的D-葡萄糖醛酸对照品溶液。
样品溶液:精密称取透明质酸钠凝胶敷料样品1 g于20 ml顶空瓶中,加入5 mol/L盐酸溶液5 mL,压盖,涡旋混匀后置于121 ℃烘箱中,加热水解1.5 h,取出冷却至室温后,用纯水全部转移至50 mL容量瓶中,加入5 mol/L氢氧化钠溶液5 mL中和,再加水稀释,定容至标线,摇匀,以0.45 μm水系滤头过滤除去沉淀物,取续滤液作为样品溶液。
PMP-甲醇溶液:称取1-苯基-3-甲基-5-吡唑啉酮8.71 g于100 mL容量瓶中,加入甲醇溶解,稀释定容至标线,摇匀,配制成质量浓度为0.5 mol/L的PMP-甲醇溶液。
空白凝胶溶液:精密称取空白凝胶1 g于20 mL顶空瓶中,与样品溶液同法制备。
1.4 实验步骤
1.4.1 衍生化反应
精密量取透明质酸钠对品溶液和样品溶液各800 μL,分别置于15 mL螺口试管中,加入0.5 mol/L的PMP-甲醇溶液0.8 mL、0.3 mol/L氢氧化钠溶液800 μL,混匀,于70 ℃恒温水浴中保温100 min,取出,冷却至室温后,加入0.3 mol/L盐酸溶液1 000 μL中止反应,混匀后,再加入二氯甲烷5 mL,涡旋萃取1 min,以3 500 r/min离心5 min,溶液分层,弃去二氯甲烷层,收集上层上清液作为衍生化样品溶液。同法制备D-氨基葡萄糖盐酸盐、D-葡萄糖醛酸的衍生化样品溶液、空白凝胶溶液和试剂空白。1.4.2 衍生物脱盐处理
将C18固相萃取小柱(柱体积为1 mL)安装在10 mL玻璃注射器上,先以5 mL的甲醇溶液冲洗小柱,接着用10 mL纯水过柱平衡,然后将透明质酸钠对照品溶液,D-氨基葡萄糖衍生化样品溶液,用纯水稀释至甲醇体积分数为1%,上样,再以20 mL超纯水冲洗小柱,用5 mL甲醇溶液洗脱并收集洗脱液,最后将洗脱液氮气吹干冷藏保存,临用前,用少量甲醇复溶后测定,用于液相色谱-质谱结构确证。
1.4.3 测定
取衍生化反应后的透明质酸钠对照品溶液及样品溶液,在1.2色谱条件下进样分析,记录色谱图峰面积,以外标法计算样品中透明质酸钠的含量。
取脱盐处理后的衍生物样品,在1.2质谱条件下进样分析,扫描衍生物的质荷比并进行结构分析及确证。
2、 结果与讨论
2.1 色谱条件的选择
2.1.1 检测波长的选择
通过二极管阵列检测器对PMP-氨基葡萄糖衍生物进行考察,图1为PMP-氨基葡萄糖衍生物特征峰的紫外光谱图,从图1中可以看出,该衍生物在219 nm波长处有最大吸收,但是该波长条件下,溶液色谱图中干扰峰较多,梯度洗脱基线波动较大。在240 nm~260 nm波段,PMP-氨基葡萄糖衍生物仍有较高响应,且峰响应值下降趋势较为平缓,因此选择250 nm作为PMP-氨基葡萄糖衍生物的测定波长,与文献[12]报道检测波长一致。
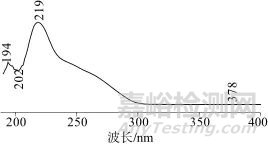
图1 PMP-氨基葡萄糖衍生物特征峰的紫外光谱图
Fig. 1 UV Spectrogram of Characteristic Peaks of Glucosamine- PMP Derivatives
2.1.2 流动相的优化
在文献[12]的基础上选择乙腈-乙酸铵缓冲盐系统作为流动相,考察了乙酸铵缓冲溶液的pH、乙腈-乙酸铵缓冲溶液的比例对PMP-氨基葡萄糖衍生物分离的影响。
表2对比了不同色谱条件下PMP-氨基葡萄糖衍生物的分离效果,图2为对应的色谱图。由表2、图2可知,在色谱条件4,即乙酸铵缓冲溶液pH用冰乙酸调节至4.5,流速为1.0 mL/min时,可改善PMP-氨基葡萄糖衍生物与相邻杂质峰的分离度,具体梯度洗脱程序见表2。
表2 不同色谱条件下PMP-氨基葡萄糖衍生物的分离效果
Tab. 2 Isolation effect of PMP glucosamine derivatives under different chromatographic conditions

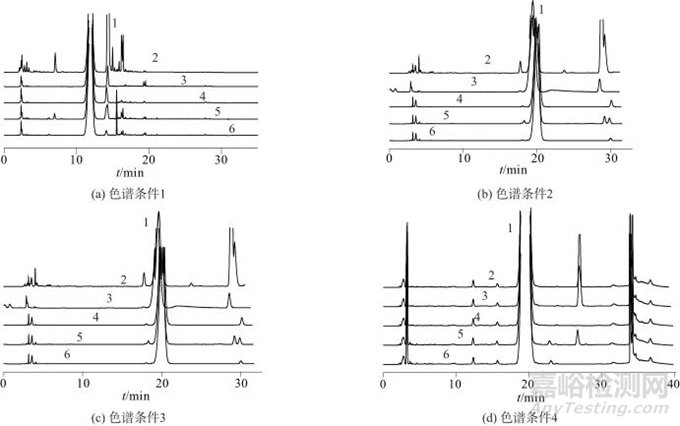
图2 不同色谱条件下PMP-氨基葡萄糖衍生物色谱图
Fig. 2 Chromatograms of PMP glucosamine derivatives under different chromatographic conditions
1—氨基葡萄糖衍生物;2—透明质酸钠对照品;3—D-氨基葡萄糖盐酸盐;4—D-葡萄糖醛酸;5—透明质酸钠凝胶样品;6—空白凝胶
2.2 透明质酸钠水解产物定性分析
透明质酸钠[11,13-15]酸水解过程:首先多糖结构的β-1,4糖苷键处发生断裂,生成二糖;第二阶段,部分二糖在高温强酸的环境下继续水解,产生氨基葡萄糖和葡萄糖醛酸;第三阶段,几乎所有二糖全部转化为单糖。在液相色谱分析中通过将D-氨基葡萄糖采用PMP标记后定位,确定了氨基葡萄糖衍生物的峰位(标记该组分为a)。图3为透明质酸钠水解物色谱图,从图3中可以看出,透明质酸钠的水解产物衍生物的峰位与PMP-氨基葡萄糖衍生物峰位一致。
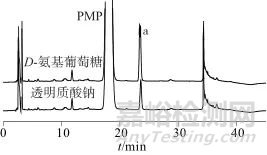
图3 透明质酸钠水解物色谱图
Fig. 3 Chromatograms of hydrolyzed components of sodium hyaluronate
为进一步确认标记组分a为PMP-氨基葡萄糖衍生物,分别收集D-氨基葡萄糖和透明质酸钠对照品水解产物的PMP衍生化溶液,采用C18固相萃取小柱对其富集、脱盐后进单级四级杆高效液相色谱质谱联用仪解析。
对标记组分a进行正负离子模式下的单级四级杆高效液相色谱质谱联用仪扫描分析,图4为PMP-氨基葡萄糖衍生物质谱图,从图4中可以看出,在负离子模式下均得到标记组分的分子离子峰质荷比均为508.2,在正离子模式下均得到标记组分的分子离子峰质荷比均分别为510.2、548.2。
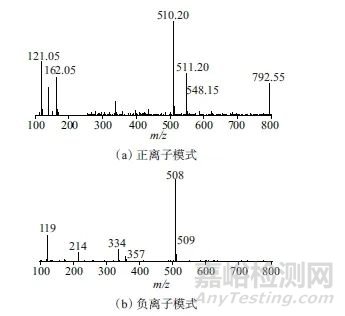
图4 PMP-氨基葡萄糖衍生物质谱图
Fig. 4 Mass spectrum of PMP glucosamine derivatives
图5为PMP与氨基葡萄糖的反应过程示意图,用于推测PMP与氨基葡萄糖的反应机理。从图5中可以看出,衍生物的分子式为C26H31N5O6,分子量为509.5,失去一个氢为[M-H]-(即m/z 508.2),加氢为[M+H]+(即m/z 510.2),加钾离子为[M+K]+(即m/z 548.2)。因此可进一步确证透明质酸钠水解产物之一为氨基葡萄糖。
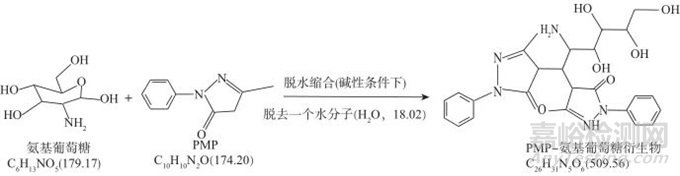
图5 PMP与氨基葡萄糖的反应过程示意图
Fig. 5 Schematic diagram of the reaction process between PMP and glucosamine
2.3 衍生化条件的优化
2.3.1 衍生化温度
采用1-苯基-3-甲基-5-吡唑啉酮(PMP)与透明质酸钠水解后产生的还原性氨基葡萄糖在碱性条件形成PMP-氨基葡萄糖衍生物,该反应需在加热条件下进行。
分别选择衍生化在50、60、70、80、90 ℃条件下进行考察温度对响应值的影响,图6为不同衍生化温度对应的色谱峰面积,从图6中可以看出,该反应从温度70 ℃起衍生化趋于平稳,因此选择衍生化温度为70 ℃。
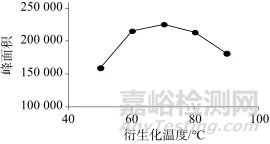
图6 不同衍生化温度对应的色谱峰面积
Fig. 6 Chromatographic peak areas corresponding to different derivatization temperatures
2.3.2 衍生化时间
考察70℃条件下加热时间对响应值的影响,分别选择衍生化时间为30、60、80、100、120 min进行实验。图7为不同衍生化时间对应的色谱峰面积,从图7中可以看出,PMP-氨基葡萄糖衍生物在80~120 min达到反应平台期,选择中间时间点100 min作为衍生化时间。
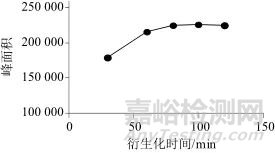
图7 不同衍生化时间对应的色谱峰面积
Fig. 7 Chromatographic peak areas corresponding to different derivatization times
2.3.3 萃取溶剂
透明质酸钠水解产物衍生化后,需要用适当溶剂将过量的PMP试剂从水相中萃取分离出去,便检测分析的同时保证衍生物的稳定,过量残留PMP试剂会引起PMP-氨基葡萄糖衍生物的降解。文献[11-12,14]多采用氯仿作为萃取溶剂,重复萃取3次后用于检测分析。氯仿为易制毒管制试剂,改用性质相近的二氯甲烷作为萃取溶剂,萃取结果见表3。由表3可知,采用两种萃取溶剂的衍生物溶液中PMP经3次萃取,萃取量达98%,二氯甲烷与氯仿对过量PMP的萃取能力一致。因此选择二氯甲烷作为萃取溶剂。
表3 二氯甲烷和氯仿萃取能力
Tab. 3 Extraction ability of dichloromethane and chloroform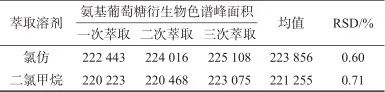
2.3.4 萃取次数
在室温条件下,分别取透明质酸钠对照品溶液、样品溶液采用二氯甲烷5 mL单次萃取,制备衍生化溶液,分别于0、10、20、30、40、48 h,注入高效液相色谱仪测定,PMP-氨基葡萄糖衍生物的响应值的相对标准偏差低于1%,表明透明质酸钠对照品衍生物和透明质酸钠凝胶敷料衍生物在室温条件下放置48 h溶液稳定性良好,衍生化溶液中存在少量的PMP试剂并不会导致衍生物降解。因此在衍生化萃取操作中可采用二氯甲烷5 mL单次萃取的方式,快速除去供试品中过量PMP,便于液相色谱分析。
2.4 水解条件的优化
2.4.1 酸浓度
透明质酸钠水解产生的氨基葡萄糖在高温浓酸的环境下稳定,且透明质酸钠对照品与透明质酸钠凝胶敷料样品水解程度一致。分别选择2 mol/L盐酸、5 mol/L盐酸、6 mol/L盐酸进行水解,结果表明,5 mol/L盐酸条件和6 mol/L盐酸条件下,对照品衍生物响应基本一致,2 mol/L盐酸条件下衍生物对照品响应值偏低,5 mol/L盐酸条件下透明质酸钠凝胶敷料样品加标回收率为98%~102%,说明5 mol/L盐酸条件透明质酸钠对照品已完全水解并且与透明质酸钠凝胶敷料样品的酸水解程度一致。因此选择最佳盐酸浓度为5 mol/L。
2.4.2 酸水解时间
考察5 mol/L盐酸条件下加热水解时间对PMP-氨基葡萄糖响应值的影响,分别选择水解时间为0.5、1.5、2.5 h进行实验。结果表明,水解时间为0.5 h时衍生物响应值偏低,水解时间为2.5 h时衍生物响应值呈下降趋势,因此选择最佳水解时间为1.5 h。
2.5 系统适用性及专属性试验
分别精密量取透明质酸钠对照品溶液、透明质酸钠凝胶敷料样品溶液、D-氨基葡萄糖溶液、D-葡萄糖醛酸溶液、空白凝胶溶液、空白试剂溶液各10 μL,在1.2色谱条件下测定。图8为该专属性试验色谱图,从图8中可以看出,样品、对照品衍生化溶液在PMP-氨基葡萄糖衍生物出峰处有相同保留时间,约为23.5 min,空白凝胶对PMP-氨基葡萄糖测定无干扰,D-葡萄糖醛酸衍生物对PMP-氨基葡萄糖测定无干扰。PMP-氨基葡萄糖衍生物色谱峰的理论塔板数大于35 000,拖尾因子为1.0,与相邻峰分离度大于1.5。
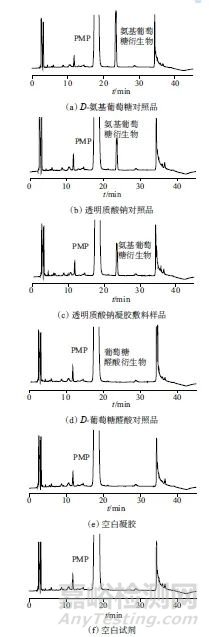
图8 专属性试验色谱图
Fig. 8 Chromatogram of specificity test
2.6 线性关系和检测限
分别将透明质酸钠对照品储备液逐级稀释,配制成质量浓度分别为20、30、40、50、60 μg/mL的透明质酸钠系列标准工作溶液。分别精密量取上述溶液,注入液相色谱仪测定,以透明质酸钠质量浓度为横坐标,以色谱蜂面积为纵坐标,绘制标准工作曲线,用最小二乘法进行线性拟合,得到线性方程y=6.531×103x-7.708×103,线性相关系数为0.999 6。结果表明,透明质酸钠的质量浓度在20~60 μg/mL范围内与色谱峰面积线性关系良好。以信噪比大于等于3计算透明质酸钠的质量分数,作为方法检出限,以信噪比大于等于10计算透明质酸钠的质量分数,作为方法定量限。得到透明质酸钠检出限为0.35 μg/mL,定量限为1 μg/mL。
2.7 精密度试验
取同一批号透明质酸钠凝胶敷料进行水解并衍生化,平行配制6份样品溶液,在1.2色谱条件下测定,记录峰面积,按透明质酸钠对照计算凝胶敷料中透明质酸钠的含量。精密度试验结果见表4。由表4可知,测定均值为2.125 mg/g,测定结果的相对标准偏差(RSD)为0.5%(n=6),表明该方法的精密度良好。
表4 精密度试验结果
Tab. 4 Results of precision test

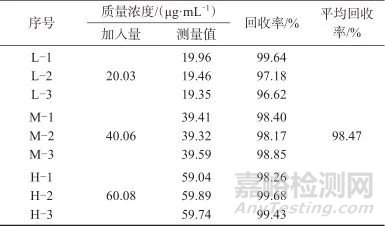
2.8 样品加标回收试验
精密称取空白凝胶1 g,置于20 mL顶空瓶中,分别加入质量浓度为20、40、60 μg/mL透明质酸钠溶液,每个浓度平行配制3份,水解和衍生化后测定,加标回收试验结果见表5。由表5可知,平均加标回收率为98.47%(n=9),表明该方法准确可靠,可用于透明质酸钠凝胶敷料中透明质酸钠含量测定。
表5 样品加标回收试验结果
Tab. 5 Results of recovery test of spiked samples
2.9 耐用性试验
分别考察同规格不同品牌色谱柱(Thermo ODS-2 C18柱、菲罗门Titank C18柱、Kromasil-100-5 C18柱)、不同柱温(±3 ℃)、波长(±2 nm)、流动相缓冲盐浓度(±10%)、流动相pH(±0.2)条件下对检测结果的影响。不同条件下PMP-氨基葡萄糖衍生物与相邻杂质峰分离度均符合要求,样品测定结果无明显差异,该方法耐用性良好。
3、 结语
通过酸水解、柱前衍生化的前处理方法的选择、分析条件的优化,最终建立了透明质酸钠凝胶敷料中透明质酸钠的检测方法。该方法专属性强,灵敏度高,应用范围广,可以满足复杂基体凝胶产品中的透明质酸钠的检测要求,为透明质酸钠各类制剂的质量控制提供参考依据。
参考文献:
1 张志舟,林嘉婷,潘灿盛,等.乙醇沉淀结合酶解—高效液相色谱法测定固体饮料中透明质酸钠[J].食品与机械,2023,39(5):64.
ZHANG Zhizhou,LIN Jiating,PAN Cansheng,et al. Optimization of extraction technology and determination of sodium hyaluronate in solid drinks by ethanol precipitation combined with enzymatic hydrolysis-reversed phase chromatography[J].Food & Machinery,2023,39(5):64.
2 王胜男,宫建辉,徐熙明,等.透明质酸钠功能机制及其在食品中的应用进展[J].食品安全导刊,2022(11):181.
WANG Shengnan, GONG Jianhui, XU Ximing,et al. The functional mechanism of sodium hyaluronate and its application in food[J].China Food Safety Magazine,2022(11):181.
3 蔡同凯,刘谋治,邓婕,等.透明质酸的作用机制及临床应用研究进展[J].药学实践杂志,2022,40(2):103.
CAI Tongkai,LIU Mouzhi,DENG Jie,et al.Research progress on action mechanism and clinical application of hyaluronic acid[J] Journal of Pharmaceutical Practice,2022,40(2):103.
4 周俊,胡犇,戢力维,等.透明质酸钠可溶微针的制备及性能特征[J].中国药业,2022,31(3):26.
ZHOU Jun,HU Ben,JI Liwei,et al. Preparation and property characterization of sodium hyaluronate soluble microneedles[J]. China Pharmaceuticals,2022,31(3):26.
5 常晓丹,王玲艳,郭独一,等.DPL联合注射用透明质酸钠复合溶液在面部年轻化治疗中的应用[J].中国美容医学,2024,33(2):77.
CHANG Xiaodan,WANG Lingyan,GUO Duyi,et al. Application effect of delicate pulse light combined with sodium hyaluronate composite solution for injection in facial rejuvenation treatment[J].Chinese Journal of Aesthetic Medicine,2024,33(2):77.
6 朱吉,王娜,耿袁,等.一种有效测量游离透明质酸钠含量的方法[J].江西化工,2024,40(1):23.
ZHU Ji,WANG Na,GENG Yuan,et al. An effective method for measuring the content of free sodium hyaluronate[J] Jiangxi Chemical Industry,2024,40(1):23.
7 朱鸿达,张松艳,陈自猷.高效阴离子离子色谱-脉冲安培法测定食品中透明质酸钠含量[J].化工管理,2023,(15):47.
ZHU Hongda,ZHANG Songyan,CHEN Ziyou.Determination of sodium hyaluronate in food by high performance anion exchange chromatography with pulsed amperometric detection[J]. Chemical Engineering Management,2023,(15):47.
8 陈玉娟,陈雯雯,乔莉苹,等.高效液相色谱法测定透明质酸钠含量的研究[J].药学研究,2020,39(3):146.
CHEN Yujuan,CHEN Wenwen,QIAO Liping,et al.Determination of sodium hyaluronate by HPLC[J]. Journal of Pharmaceutical Research,2020,39(3):146.
9 张莉,赵鹏,何涛,等.高效凝胶色谱法同时测定眼用粘弹剂中透明质酸钠和硫酸软骨素钠的含量[J].理化检验(化学分册),2018,54(3):4.
ZHANG Li,ZHAO Peng,HE Tao,et al. Simultaneous determination of sodium hyaluronate and sodium chondroitin sulfate in eye viscoelastic agents by high performance gel chromatography[J]. Physical Testing and Chemical Analysis(Part B:Chemical Analysis) ,2018,54(3):4.
10 巩婉秋,王静怡,朱雨,等.透明质酸钠含量近红外检测方法研究[J].中国检验检测,2023,31(5):33.
GONG Wanqiu,WANG Jingyi,ZHU Yu,et al. Research on near-infrared detection method for sodium hyaluronate content[J]. Chinese Journal of Inspection and Testing,2023,31(5):33.
11 黄芳,邓欣,王玉芹,等.酸水解-柱前衍生化-高效液相色谱法测定面膜化妆品中透明质酸[J].理化检验(化学分册),2019,55(8):898.
HUANG Fang,DENG Xin,WANG Yuqin,et al. Determination of hyaluronic acid in facial mask cosmetics by acid hydrolysis pre column derivatization high performance liquid chromatography[J]. Physical Testing and Chemical Analysis(Part B:Chemical Analysis) ,2019,55(8):898.
12 曹辰辰,郑昕,SUN-WATERHOUSE Dongxiao等.同时测定软骨酶解物中硫酸软骨素及透明质酸含量的高效液相色谱法的建立[J].现代食品科技,2023,39(5):281.
CAO Chenchen,ZHENG Xin,SUN-WATERHOUSE Dongxiao ,et al. Establishment of high performance liquid chromatography for determination of chondroitin sulfate and hyaluronic acid in cartilage hydrolysate[J]. Modern Food Science and Technology,2023,39(5):281.
13 张璐瑶,赵峡,陈欢欢.糖类化合物PMP衍生分析进展[J].分析测试学报,2016,35(3):6.
ZHANG Luyao,ZHAO Xia,CHEN Huanhuan. Advance in 1-phenyl-3-methyl-5-pyrazolone(PMP)derivatization analysis of carbohydrates[J] Journal of Instrumental Analysis,2016,35(3):6.
14 祝贺.高效液相色谱鉴别糖胺聚糖种类方法的建立及其应用[D].山东:中国海洋大学,2014.
ZHU He.A method to distinguish glycosaminoglycans by high performance liquid chromatography and its applications[D]. Shandong:China Ocean University,2014.
15 张萍,王仲孚,黄琳娟.糖类物质的衍生化试剂—氘代PMP和类似物的合成及其衍生化性能[J].合成化学,2013,21(3):262.
ZHANG Ping,WANG Zhongfu,HUANG Linjuan.Saccharide's Derivatization Reagent—Synthesis and derivatization performances of deuterium PMP and its analogues [J].Chinese Journal of Synthetic Chemistry,2013,21(3):262.
16 黄春跃,杨子璇,孙宜春,等.基于PMP柱前衍生化-HPLC法的淫羊藿多糖的单糖指纹图谱研究[J].中国医药工业杂志, 2024,55(1):81.
HUANG Chunyue, YANG Zixuan, SUN Yichun,et al. Monosaccharide fingerprint of epimedium polysaccharides based on PMP pre-column derivatization and HPLC method[J] Chinese Journal of Pharmaceuticals, 2024,55(1):81.
17 刘永玲,赵建国,赵治兵,等.PMP柱前衍生HPLC法测定八月瓜果皮多糖中单糖组成[J].食品研究与开发,2023,44(21):131.
LIU Yongling,ZHAO Jianguo,ZHAO Zhibing,et al. Determination of monosaccharide composition of polysaccharides from akebia trifoliate peel by PMP-HPLC with pre-column derivatization[J]. Food Research and Development,2023,44(21):131.
18 孙红梅,李明华,程显隆,等.柱前衍生化高效液相色谱法测定麦冬中多糖的含量[J].中国现代中药,2022,24(11):2 126.
SUN Hongmei, LI Minghua, CHENG Xianlong,et al.,Determination of polysaccharides in ophiopogon japonicus by pre-column derivatization high-performance liquid chroma-tography[J].Modern Chinese Medicine,2022,24(11):2 126.

来源:化学分析计量


