您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2025-01-20 15:55
上周,美国食品和药物管理局(FDA)发布了上市前批准申请(PMA)和人道主义器械豁免(HDE)模块审查的最新指导文件。
本指导文件的目的是向行业和 FDA 工作人员提供有关上市前批准申请(PMA)和人道主义设备豁免(HDE)模块审查计划的信息,并概述提交或审查模块 PMA 或 HDE 的程序。这一指南从2003年11月3日首次发布以来,经过多次修订,以适应不断变化的监管需求和行业实践。
指南详情可以参考以下网址:https://www.fda.gov/regulatory-information/search-fda-guidance-documents/premarket-approval-application-and-humanitarian-device-exemption-modular-review
1、模块化审查
模块化审查方法旨在提供一种机制,申请人可以通过该机制提交非临床数据和制造信息进行审查,同时仍然使用最终设备设计收集、编译和分析临床数据。因此,模块化PMA或HDE是在不同时间提交的部分或“模块”的汇编,它们一起成为一个完整的应用程序。这种方法的目标是通过允许申请人在完成非临床测试和分析后不久向FDA提交申请的离散模块进行审查,从而提高审查过程的效率。此外,与传统的PMA或HDE申请相比,模块化方法允许申请人在审查过程中尽早解决FDA注意到的任何缺陷。
2、适用范围
模块化审查程序是传统PMA和HDE的准备、提交和评估的替代方案。由于FDA认为PMA和HDE补充材料很少适合进行模块化审查,因此本文件的范围仅限于寻求批准的初始或早期PMA和HDE的申请人。话句话说,对于接近提交完整PMA或HDE申请,或设备设计尚不稳定的产品,模块化审查可能不太适用。
3、关键词定义
➤ 壳文件(Shell):构成PMA或HDE各模块内容的概要和描述,包括各模块的提交时间表和具体计划。
➤ 模块(Module):PMA或HDE的每一个独立的审查单元,可以单独提交和审查。模块是一组元素、测试、信息等,针对医疗器械申请的特定领域或信息(例如,生物相容性和工程台架测试)。
➤ 模块修正(Module Amendment):对已提交模块的修改或补充。例如,申请者对缺陷信的回应或更新的联系信息可以作为模块修正案提交。
4、提交模块化PMA或HDE的步骤
STEP1:联系审核小组
如下附录I包括模块评审中通常涉及的步骤的流程图。第一步应该是通过电子邮件联系CDRH/CBER。
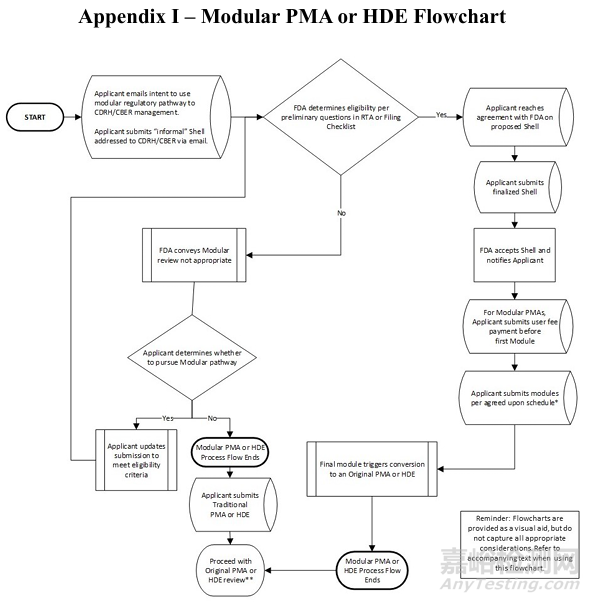
STEP2: PMA或HDE壳文件的内容与提交
a. 壳文件应包含各模块的详细内容和提交时间表,确保涵盖21 CFR 814.20(PMA)或21 CFR 814.104(HDE)要求的所有要素。
b. 企业应与FDA审查团队互动,达成壳文件的共识后,正式提交壳文件。FDA将在15天内确认接受。
STEP3: 模块的提交与审查
a.每个模块应包含封面信、目录、执行摘要、设备描述、参考文献等,并明确标识为模块提交。
b.FDA承诺在90天内完成每个模块的审查,并发出缺陷信或接受通知。模块之间应保持合理的提交间隔,通常建议至少90天。
c.若模块不完整,FDA将发出缺陷信,待补充完整后重新启动审查时钟。
STEP4: 最终模块的提交与审批转换
a.最终模块应包含临床数据、拟议标签、安全性和有效性总结等,并整合所有先前模块的内容。
b.提交最终模块后,FDA将模块化申请转换为完整的PMA或HDE,并进行接受/备案审查。PMA的接受决定在15天内,备案决定在45天内;HDE的备案决定在30天内。
5、收费与支付
➤ 收费
对于模块化PMA,首模块提交时需缴纳用户费用。若首模块提交时未支付费用,FDA将不予接受。
➤ 退款
模块化PMA的退款政策与传统PMA不同,需参照FDA相关指导文件。
➤ HDE申请不涉及用户费用,但仍需遵循模块化审查的流程。
模块化审查途径为PMA和HED医疗器械申请者提供了一种更灵活的提交方式,有助于在产品开发早期阶段就开始与FDA进行沟通和审查,从而提前发现并解决潜在问题。这必将提高审查效率和拿证的成功率。

来源:Internet


