您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-09-22 16:25
1、什么是除菌过滤,什么是除菌过滤器?
依据国家药监局2018.10.1生效版《除菌过滤技术及应用指南》中的定义:除菌过滤是指采用物理截留的方法去除液体中的微生物,以达到无菌药品相关质量要求的过程。
除菌级过滤器指在工艺条件下每平方厘米有效过滤面积可以截留大于等于1×107cfu的缺陷型短波胞菌(Brevundimonasdiminuta,曾用名:缺陷假单胞菌)的过滤器。具体可参考ASTM 838-15,使用缺陷型短波胞菌对除菌级过滤器进行挑战,以确认除菌过滤器的微生物截留能力。
2、除菌过滤器产品为什么要做验证?
主要基于药品(产品)与过滤器之间的相互影响,有来自于产品润湿后的完整性试验,以及过滤器对产品的溶出物(可提取物或浸出物)试验,吸附试验,还有产品对过滤器细菌截留率方面的影响。这些影响对应需要做相关的试验来验证。
3除菌过滤器可参考的法规有哪些?
除菌过滤器可参考的相关验证的法规如下:
US FDA 21 CFRPART 211.65
FDA Guideline onSterile Drug Products Produced by Aseptic Processing Sep. 2004
EU GoodManufacturing Practices (2016 draft) and the Annex 1: Manufacture of Sterile Products (2020 draft)
国家药品监督管理局GMP无菌药品/无菌药品附录 (2010 版)
InternationalStandard ISO 13408-2 Part 2: Filtration (2018)
PDA TechnicalReport No.26-Sterilizing Filtration of Liquids, Revised 2008
灭菌无菌工艺验证指导原则sterile, aseptic process validation guideline
国家药品监督管理局《除菌过滤技术及应用指南》(2018.10)
《化学药品注射剂灭菌和无菌工艺研究及验证指导原则》(2020.08)
4、除菌过滤器验证内容有哪些?
依据国家药监局2018.10.1生效版《除菌过滤技术及应用指南》中5.1除菌过滤验证概述,表明除菌过滤器验证内容包括:性能确认(由过滤器厂商-依据药典方法进行)和过滤工艺验证(由过滤器用户或委托第三方实验室经评估来设计试验)两部分。下图展示出性能确认和过滤工艺验证对应的项目。
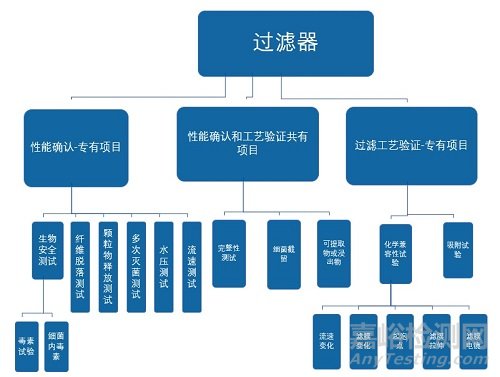
5、除菌过滤器的过滤工艺验证具体有哪些?
依据国家药监局2018.10.1生效版《除菌过滤技术及应用指南》中5.1除菌过滤验证概述,过滤工艺验证是指针对具体的待过滤介质,结合特定的工艺条件而实施的验证过程,一般包括细菌截留试验、化学兼容性试验、可提取物或浸出物试验、安全性评估和吸附评估等内容。如果过滤后,以产品作为润湿介质进行完整性测试,还应进行相关的产品完整性测试验证。除菌过滤工艺验证可以由过滤器的使用者或委托试验检测机构(例如:过滤器的生产者或第三方试验室)完成,但过滤器使用者应最终保证实际生产过程中操作参数和允许的极值在验证时已被覆盖,并有相应证明文件。
一、完整性测试:应明确过滤器使用后完整性测试的润湿介质(包括谁、乙醇等标准介质或药液)。如果采用的润湿介质为药液,则应进行产品相关完整性标准的验证以支持该标准的确定。完整性测试贯穿于细菌截留试验以及化学兼容性试验,除此之外,在药品连续三批生产工艺验证及常规生产过程中也涉及完整性测试。(来源于化学药品注射剂灭菌灭菌/无菌工艺研究及验证指导原则。)
二、细菌截留:细菌挑战试验的研究目的是模拟实际生产过滤工艺中的最差条件,过滤含有一定量挑战微生物(根据ASTM 838-15,使用缺陷型短波胞菌(Brevundimonas diminuta,曾用名:缺陷假单胞菌)对除菌级过滤器进行挑战的产品溶液或者产品替代溶液,以确认除菌过滤器的微生物截留能力。(来源于国家药监局2018.10.1生效版《除菌过滤技术及应用指南》章节5.2)。
三、可提取物或浸出物:可提取物试验在选择模型溶剂之前必须对产品(药品)处方进行全面的评估。用于测试的模型溶剂应能够模拟实际的药品处方,同时与过滤器不应有化学兼容性方面的问题。可以从过滤器及其他组件材料的工艺介质接触表面提取出的化学物质。应先获得最差条件下的可提取物数据,将其用于药品的安全性评估。可提取物反映了浸出物的最大可能。除菌过滤器的浸出物研究,需要根据提取研究结果和风险评估结果确定。一般可提取物和浸出物试验的研究策略,是在可提取物评估基础上,针对高风险的组件进行进一步的浸出物研究。应针对过滤器可提取物或浸出物的种类和含量,结合药品最终剂型中的浓度、剂量大小、给药时间、给药途径等对结果进行安全性评估(参考化学药品注射剂生产所用的塑料组件系统相容性研究技术指南,一般使用SCT/TTC/QT/PDE进行评估,以评估可提取物和浸出物是否存在安全性风险。(来源于国家药监局2018.10.1生效版《除菌过滤技术及应用指南》章节5.3)。
四、化学兼容性试验:过滤器化学兼容性说明该工艺流体及其工艺条件对该过滤器的特定性能没有产生负面影响。通常, 过滤器的性能包括过滤器的物理强度(在一定温度下, 能耐受一定的压差)、允许流体通过的能力(即通透性,如流速和通量)和去除流体中粒子的能力(即截留性能,如细菌、病毒或颗粒)。
化学兼容性试验检测项目一般有:过滤器接触待过滤介质前后的目视检查;过滤过程中流速变化;滤膜重量/厚度的变化;过滤前后起泡点等完整性测试数值的变化;滤膜拉伸强度的变化;滤膜电镜扫描确认等。
PDA 26号第4.1章节:提到完整性测试和细菌挑战测试的结果可以用来评估化学兼容性。
五、吸附试验:吸附是所过滤的料液中的某些成分粘附在滤膜上的过程,可能影响料液的构成和浓度。过滤器中吸附性的材料包括滤膜、硬件和支撑性材料。吸附试验条件可以根据实际生产条件确定,流速、过滤时间、料液浓度、防腐剂浓度、温度和pH 值等因素都可能影响吸附效果。吸附试验中采用的检测方法可以采用产品质量标准中所确定的相关检测方法。(来源灭菌无菌工艺验证指导原则sterile,aseptic process validation guideline,3.2.2.4吸附试验)。

来源:NSF认证检测


