您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-08-10 21:12
一款新药从研发到上市都需要经过哪些流程?每一步又有哪些经验可以借鉴?本文以小分子药物为例,试着做了一个梳理,希望能对您有所帮助。
一、临床前研究
1.研究开发(一般 2-3年)
实验室研究,寻找治疗特定疾病的具有潜力的新化合物
1 )药物靶点的发现及确认
这是所有工作的起点,只有确定了靶点,后续所有的工作才有展开的依据。
2)化合物的筛选与合成
根据靶点的空间结构,从虚拟化合物库中筛选一系列可匹配的分子结构,合成这些化合物,它们被称为先导化合物。
3)活性化合物的验证与优化
不是所有先导化合物都能符合要求,在这个阶段需要通过体外细胞试验验证,初步筛选出活性高、毒性低的化合物,并根据构效关系进行结构优化,这些化合物称为药物候选物。
同时也存在一个化合物对目标 A 靶点没有作用,却有可能对其他的 B 靶点、C 靶点有非常好活性的情况,暂且不表。
2.临床前实验(一般 2-4年)
这一阶段目的,一是评估药物的药理和毒理作用,药物的吸收、分布、代谢和排泄情况(ADME)。二是进行生产工艺、质量控制、稳定性等研究(CMC)。
第一部分的实验需要在动物层面展开,细胞实验的结果和活体动物实验的结果有时候会有很大的差异。这一步的目的是确定药物的有效性与安全性。
第二部分需要在符合GMP要求的车间完成。
1 )药理学研究
包括:药效学、药动学
2 )毒理学研究
急毒、长毒、生殖毒性,致癌、致畸、致突变情况
3 )制剂的开发
总不能弄点化合物就直接往嘴里倒吧,制剂开发是药物应用的一个重要环节。比如有的药口服吸收很差,就需要开发为注射剂。有的药在胃酸里面会失去活性,就需要开发为肠溶制剂。有的化合物溶解性不好,这也可以通过制剂来部分解决这个问题。还有的需要局部给药,就需要通过制剂开发成雾化剂、膏剂等。
二、临床试验审批 Investigational New Drug(IND)
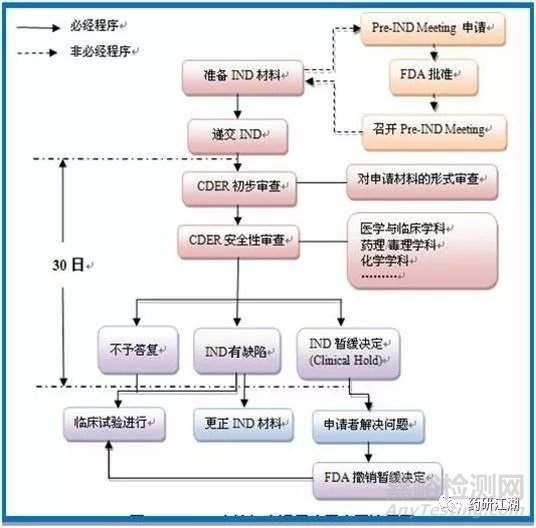
三、临床试验(一般3-7年)
人体试验共分三期:
• Ⅰ期临床 20-100例,正常人,主要进行安全性评价。
• Ⅱ期临床 100-300例,病人,主要进行有效性评价 。
• Ⅲ期临床 300-5000例,病人,扩大样本量,进一步评价。
因为 Ⅰ-Ⅲ 期临床在整个药物研发过程中非常重要,我们重点讲一下这部分。
传统意义上,新药的临床研究分为I/II/III期,后来 II 期有分成 IIa 和 IIb(很大程度上是因为肿瘤药物研究),接着出现了 0 期研究的概念(很大程度上也是因为肿瘤药物的研发)。然后又有人提出,0/I 期为早期临床研究,IIa 为中期临床研究,IIb 和 III 期为晚期临床研究。
这种分类,其实我觉得在两个方面对我们进行临床研究设计的人来说,是很重要的:
1. 分期是和研究目的相关的,不同的目的,可以分到不同时期/阶段的。
2. 不同时期/阶段的临床研究,往往对应于一定的研究框架设计(交叉研究,平行对照,单臂,终点选择等等)。
所以,理解不同时期/阶段的临床研究,其意义已经超出了知道有这样的分法本身。
分期的一头是研究目的,另一头是对应的设计框架(当然也只是粗略的框架,很多问题,要根据疾病、药物等来确定),理解分期,有助于我们了解,一个研究目的,大体上有哪些设计方法和思路,大体上可以如何去处理;为什么该如此进行,各种设计的优缺点在哪里。
所以,我们先看一下不同阶段的研究目的。
1. 0 期
目的是在满足一定的统计学要求的前提下,在有限的样本量(通常不超过20)、有限的时间内(每个患者的治疗期常不超过数周),初步判断一个研究药物是否有效、某个剂量是否有效,是否应继续开发下去,附带看一下某些疗效判断方法是否可行。
从目的角度,0 期研究也是初步判断药物的效果,类似 II 期(尤其是IIa),所以 0 期的设计,在某些方面类似 IIa 的单臂研究,但是由于样本量小、又要满足一定的统计学要求,所以也有其特点,设计起来其实可能更为复杂。
2. I 期
通常是摸索剂量(dose finding, dose-ranging),药代(PK)和药动(PD),样本量也不大(一般不超过20,或者20左右)。尤其是PD,通常会使用交叉研究,因为交叉研究有其特点,适合此类设计目的。
交叉研究中,每个患者都是其自己的对照(在不同阶段,分别扮演研究组和对照组的角色),而且交叉研究的变量一般小于平行对照,所以在利用样本方面,交叉研究的效率远高于平行对照,交叉研究如果纳入 20 例患者,平行对照 50 例都不一定能获得相似的结果。
但是交叉对照也有很多限制,例如脱落和失访对交叉研究的影响远大于平行对照研究;同一个人,不同时期接受不同治疗、中间隔以洗脱期,导致后遗效应,以及治疗-时间相互作用等等。所以在后期临床研发中,很少看到有用交叉对照的(有,但远少于平行对照)。
3. II 期
II 期是初步判断疗效的和安全性的(其实安全性贯穿研发始终),所以一般称为safety & activity研究(SA研究)。有多种做法,根据不同的临床疾病环境,以及既往数据等,可以采取单臂,平行对照的 RCT 等,终点一般选择 surrogate,而不是 clinical outcome。如果分 IIa 和 IIb,则 IIa 往往是单臂,样本量一般不会过百;IIb 则往往是平行 RCT。
4. III 期
III 期是严格的验证药物效果的验证性临床研究。很多会议上介绍临床研究,往往以此类研究为模版进行介绍,在假设检验的框架下进行介绍。
四、新药上市审批 New DrugApplication
●NDA申报资料 — CTD(CommonTechnical Document)
CTD主要由五大模块组成:
①行政和法规信息
②概述:药物质量、非临床、临床试验的高度概括
③药品质量详述
④非临床研究报告
⑤临床研究报告
流程:
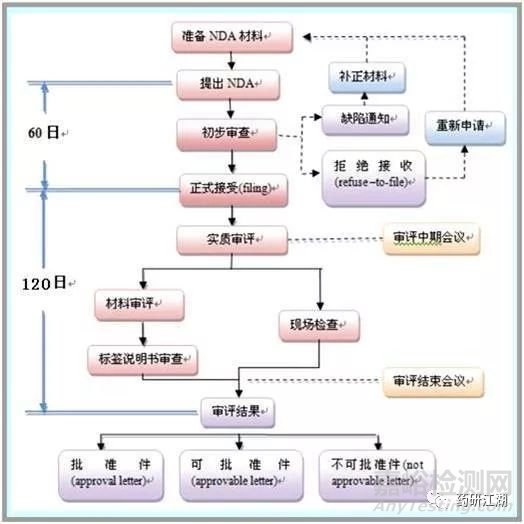
①批准信
符合要求,可以上市
②可批准信
基本满足要求,少数不足可以修改 。
申请人应在收到10日内作出回应修正,否则视为自动撤回。
③拒绝信
存在严重问题或需要补充大量信息资料。
申请人可在10日内提出修正或在30日内要求听证
●NDA特殊审评程序
①优先审评(PriorityReviews)
适用于能够在治疗、诊断或预防疾病上比已上市药品有显著改进的药品,优先安排NDA审评。
②加速审批(Accelerated Approval)
用于治疗严重或危及生命疾病的药品,且存在合理并能够测量的“替代终点”(Surrogate endpoint),即药物预期的治疗效果的指标,变通审评标准,利用“替代终点”审评。
③快速通道(Fast-track)
用于治疗严重或危及生命疾病的药品,且有潜力满足临床尚未满足的医学需求,早期介入,密切交流,分阶段提交申报资料。
以万络为例, 1998 年 11 月 23 日万络提交 NDA 申请,编号 21-042,“1999 年 5 月 20 日获 FDA 批准,历时 178 天。
五、上市后研究
临床监测期:IV期临床
受试者要大于 2000 例,同时要进行社会性考察。
仍以万络为例:2000 年进行了“VIGOR”胃肠道试验 ——显示较少的胃肠道副作用, 但是使用 18 个月后会引发 2 倍的心脏病/中风风险。
2001 年,“APPROVe”腺瘤息肉预防试验 ——服药超过 18 个月出现较高的心血管疾病风险。
六、上市后再审批(一般上市后4-10年)
目的:重新审核 NDA 中的有效性和安全性。
万络,2002 年 4 月:默克公司增加了万络可能出现心血管副作用的警告。
2004 年 9 月 28 日,默克公司与 FDA 商讨有关万络实验结果的事宜。
2004 年 9 月 30 日, 再审评:“万络”,由默克公司主动召回。

来源:Internet


