您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-03-15 22:47
在所有生物制药行业中,清洁验证始终是一个非常重要的工作。最近几年,生物制药正在中国快速发展,早期几年上市一个产品,到现在一年很多产品上市;从一个企业只有一个品种,到多产品共线生产;从融合蛋白到单抗、双抗、多抗、ADC,以至溶瘤病毒、CAR-T、CAR-NK等各类生物技术多点开花,生物技术产品的生产企业在现有的市场条件下各自冲击不同赛道,更有很多的CDMO企业快速发展。
这些快速的发展,在早些年甚至对监管和合规都带来了一定的挑战。近几年,国家层面出台大量法规、指南用于指导企业和监管部门,进而有效地推动生物制药行业的快速发展。但对于生物制品清洁相关的法规和指南仍然偏少,在笔者的工作实践中,经常发现在企业中很多人并不清楚该如何有效的开展清洁验证,生物制品有很明显的特点:
工艺过程复杂,自种子复苏到原液,通常有10几个步骤,并且有培养基配制、十几种以上的缓冲液配制和储存过程。
工艺周期较长,通常都在一个月以上的时间。
工艺设备种类多,常见的生物制品厂用到的直接工艺设备就有几十种,罐体可能就有70多个,而且还有越来越多的一次性设备和系统的使用。
原材料种类繁多,仅培养基中的物质可能就达40多种,且不算十几、二十几种的缓冲液。
早期的目标产品(如蛋白)并不是主要的污染物,如生物反应器阶段,基质细胞及碎片、培养基物质的浓度可能比目标蛋白更多。
随着产品向下游移动,对患者的影响也越来越明显且直接。
某些污染物并不是最后一步才去除(如宿主蛋白)。
生物制品的各种物质的活性通常都非常高,如果采用化药的算法,允许残留限度可能会非常非常低,以致于清洁工艺可能无法实现。
对残留影响最大的不一定是目标物质,目标蛋白可能随着清洁和灭菌程序就会失活、变性、分解而变得不存在了。
鉴于上述特点,关于生物制品清洁验证中的问题或错误主要包括以下几个方面:
在早期工艺开发的过程中未能同步开展清洁工艺和用于清洁验证的分析方法的开发和确认,以至于在上市申报的工艺验证时同步开展清洁验证工作时,无法确定包括清洁工艺、残留限度等各项参数,少数企业仍有将验证当作工艺开发对待的情况,以期望在清洁验证过程中完成清洁工艺开发。
在新厂建设过程中,对CIP、SIP、离线清洗、手动清洗过程的定义不清楚,存在过度设计(如所有的不锈钢管罐均在每次使用后进行SIP)和设计不足(如离线清洗、废物灭活的种类和数量计算不足)等情况。
设备的设计和选型未充分考虑清洁验证和日常监测取样点的设置。
对取样和检验方法的考虑不充分,取样点设计未体现最差条件,检验方法选择不合适(如无机盐类的缓冲液的残留使用TOC作为残留指示)。
在清洁验证残留限度设计过程中全线工艺采用一个标准,导致上游达不到,下游标准过低,受到监管挑战。
在残留限度制定时,下游的残留限度高于上游,制定的残留限度标准甚至严于清洁用水的标准,如内毒素标准为0.06 EU/ml。
取样点不同,企业在清洁验证过程中存在两个极端,有些公司的清洁验证擦拭取样点多达3000个,而有些公司的取样只有冲淋水而无擦拭取样。
检验用分析方法未经过确认或验证,如使用常规浓度检测方法检测清洁后残留限度,超方法验证范围使用。
在制定脏设备保留时间(DHT)时过短以致于岗位操作人员无法实施,或者时间过长,以致于清洁取样回收率都无法保证。干净设备保留时间(CHT)也同样存在过短或过长的情况。
清洁过程无维护,清洁过程无监测,只能通过再验证的模式进行验证状态维护。
整体方案无统筹,对于标准制定、取样计划、取样方式、检测方法、清洁工艺稳健性的证明所需数据考虑不充分,哪些是小试中试过程中的研究数据,哪些必须在大生产进行清洁验证均有所缺失。
编写《清洁验证主计划》时套用模板,与公司实际情况不一致。
上述问题或缺陷并非来自同一家公司,而是众多公司中较为普遍的一种现象,正是因为生物制品的特点和由于整个行业快速发展导致的人才相对的匮乏,才出现的上述的很多问题,综合上述问题和错误,归纳起来笔者认为是以下几点原因:
对法规指南不熟悉;
在工艺开发过程中仅注重了工艺路线的研究而忽视了辅助路线的开发;
对风险认识不到位,过严和过松都是认知不正确的体现;
经验和知识欠缺。
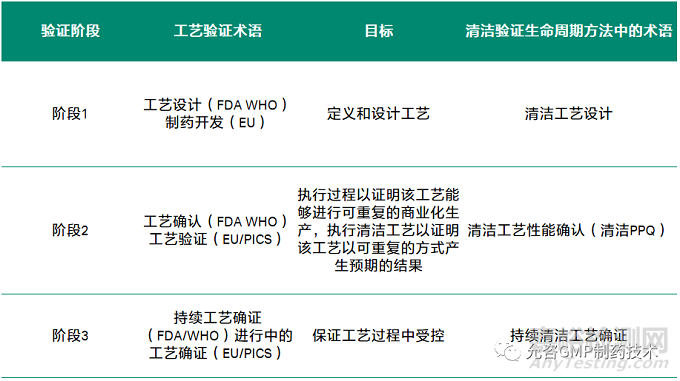
现有对于生物制品清洁验证的可参考的指南和法规中,PDA技术报告49《Points to Consider for Biotechnology Cleaning Validation生物产品清洁验证考虑要点》(2010年)依然是最具权威性和技术性的参考资料,ISPE在2020年《Cleaning Validation Lifecycle -Applications, Methods, and Controls》则正式提出了类似于工艺验证三个阶段的模式,将清洁验证分成了清洁工艺开发、清洁工艺确认和持续清洁工艺确证三个阶段。
鉴于PDA 技术报告49诞生于2010年,彼时生命周期的概念在制药行业刚刚提出,因此建议将上述两个指南结合考虑更为合适,ISPE的指南结合了三个阶段的生命周期的概念,但并不专属于生物制品,而PDA技术报告49则更针对于生物制品清洁工艺的相关要求。
总结
清洁验证的基本理念,即全生命周期、基于风险和质量源于设计的清洁验证,清洁验证的整体思路可以总结为下图所示方式:
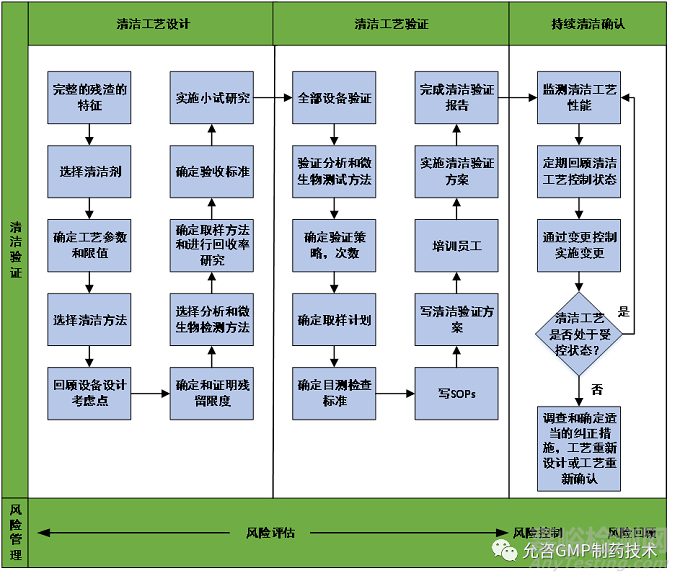
此图来源于ISPE Cleaning Validation Lifecycle -Applications, Methods, and Controls(2020),Figure 1.3: Cleaning Process Validation Road Map
上图借用了US FDA Guidance for Industry: Process Validation: General Principles and Practices, January 2011中的全生命周期的概念,对比的考虑的方式如下:
根据上述图标,清洁验证按照流程可以转换为另一种图形:
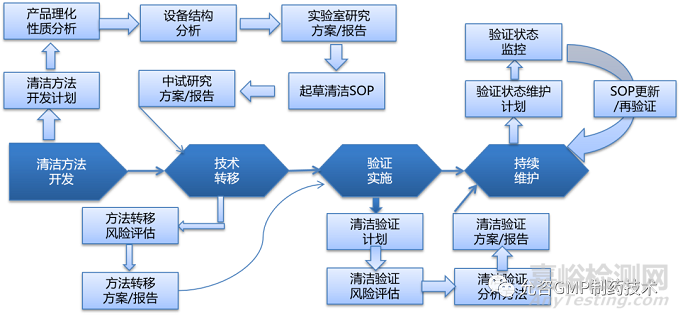

来源:允咨GMP制药技术


