您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-04-04 17:54
小编和大家一起继续学习EMA亚硝胺杂质问答的后续部分。
10. 亚硝胺杂质限度
下表限度是仅限于含有单一亚硝胺杂质的药品。
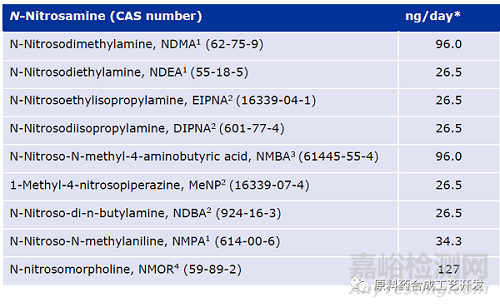
新的单一亚硝胺杂质限度计算
如果有充足的特定动物致癌性数据,根据TD50数据,按照ICH M7指导原则,计算物质特定的终身暴露限度。
如果没有充足致癌数据,以TTC不得过18ng/day计算限度。
多个亚硝胺杂质限度的计算
检出的所有亚硝胺总日摄入量,不得过所检出效力最高的亚硝胺杂质的AI值。确认的亚硝胺杂质可接受限度AI。
检出的所有亚硝胺计算所得总风险水平不超过1/100000风险。
11. 如果在制剂中检出亚硝胺,该如何做?
不管检出量多少,只要在产品中发现亚硝胺杂质,MAH/申请者就要通知当局。
以ng或者ppm单位提供报告结果,同时提供检验方法,亚硝胺杂质限度的计算方法,包括最大日剂量等详细信息。
如果发现的亚硝胺杂质低于可接受限度或者多个杂质总风险水平不超过1/100000理论终生癌症风险。MAH控制药品中亚硝胺杂质低于或者等于可接受限度,采取措施缓解亚硝胺杂质形成和污染风险。
如果单个或者多个亚硝胺杂质超过了可接受限度,MAH应该立即提交调查报告,包括根本原因,风险缓解计划和效益/风险评估,当局评估风险平衡,并采取行动。
如果特定的亚硝胺杂质已经报告当局,限度低于可接受限度,不需要再报告。
12. 亚硝胺风险有什么缓解措施?
药品中存在的亚硝胺杂质应该被尽可能的降低。无论什么时候,MAH都应该设计和调整生产工艺去阻止亚硝胺杂质形成和污染。
MAH应该执行亚硝胺杂质控制策略,包括当前和未来的措施去降低亚硝胺杂质产生和污染的风险。措施包括工艺改变,物料质量改变,质量标准建立,设备清洁验证,合理方法开发验证等。
MAH控制亚硝胺水平低于可接受限度,以应对未来供应商变更,工艺变更,包装变更带来的风险。也要确保用于制备药品的原料药和辅料的生产符合GMP。
13. 上市许可要求进行哪些改变?
MAH应该介绍API或者药品的变化,包括生产工艺,质量标准,控制策略,原材料和包装材料。当亚硝胺杂质已经确认,药品中亚硝胺杂质的质量标准中限度也要介绍。
14. 新的和正在审评的上市许可申报(MAA)处理方法如何?
申报阶段
申请者应该以附件的形式在模块1中提交风险评估,并在申报资料3.2处引用。
如果亚硝胺杂质风险存在,申请者应该提交影响药品和风险缓解策略平衡的评估,申请者应该提供确认性检测计划和检验数据。
上市许可审评阶段
如果MAA中没有提交风险评估,在MA审核阶段要求提供。如果申请者风险评估不充分,MA阶段要求进一步对亚硝胺杂质存在风险进行评估。
对于新的,正在审评的MAA,必须充分了解亚硝胺杂质来源,并提供六批中试或三批生产批次数据作为确认性检验。
如果亚硝胺杂质形成风险靠近药品,更多批次要求检验,如果使用多个生产商的物料,需要额外批次进行检验。
15. 什么时候亚硝胺杂质检验应该包含在MA档案里?
当亚硝胺杂质风险存在,需要进行确认性检验时,药品的质量标准应该包含亚硝胺杂质限度,同时药品符合限度要求。
如果亚硝胺杂质来源没有弄清楚,药品需要常规检测。
亚硝胺杂质的控制点应该能保证亚硝胺杂质存在低于可接受的限度。通常在药品中检验亚硝胺杂质,但是如果亚硝胺杂质源在API工艺阶段,可以在API中控制,以说明药品中亚硝胺杂质符合限度。即使控制点在API阶段,药品质量中也应该包含亚硝胺杂质限度,同时批次检验结果符合限度。
期望常规检测,但是如果充分理解亚硝胺杂质的根本原因,可以不常规检验。如果亚硝胺杂质存在低于可接受限度的10%,可以不检验,如果亚硝胺杂质存在低于可接受限度的30%,可以根据ICH Q6A进行跳检。
16. MAH对具有CEP或ASMF的API负有什么责任?
MAH/申请人,生产许可持有人,API生产商应共同努力,采取预防策略,降低化学合成API的药品生产和贮存过程产生亚硝胺的风险。
17. 沙坦类药品中亚硝胺杂质的第5(3)款转引评估结果的教训总结是怎样的?
和我们实际应用关系不大。
18. 其它地区的监管要求是怎样的?
和我们实际应用关系不大。

来源:原料药合成工艺开发


