您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-02-27 09:03
摘要
元素杂质的研究和控制对于保障药物的质量和安全性具有重要意义,国际人用药品注册技术协调会(ICH)已发布实施ICH Q3D指南作为药物中元素杂质研究和控制的全球协调统一的指南。基于 ICH Q3D 指南对元素杂质风险评估和控制的要求,元素杂质检测方法的开发和验证是分析工作者的关注重点。该文主要综述电感耦合等离子体原子发射光谱法(ICP-AES)和电感耦合等离子体,质谱法(ICP-MS)方法开发的要点,包括确定待测元素种类和限度、前处理方法选择、干扰和校正:具体分析ICH Q2(R2)与各国药典通则关于元素杂质测定方法的验证要求,并详细对比说明各项验证试验的评价方式,为元素杂质检测方法的开发和验证提供参考,同时也为相关研究工作者提供研究思路。
【关键词】元素杂质;ICH Q3D;电感耦合等离子体原子发射光谱法;电感耦合等离子体-质谱法
药物的杂质分为有机杂质、无机杂质和残留溶剂,无机杂质中一个重要的类别为元素杂质。一般情况下元素杂质不能为患者提供任何治疗作用,而且由于某些元素杂质具有毒性,积累到一定量后可能引起健康风险,另有些元素可能影响药品的稳定性,对药品的有效期产生不利影响。为了保证患者用药安全,以及药品的质量可控性,需对元素杂质进行风险评估与控制.
国际人用药品注册技术协调会(The Intemmationa Councilfor Harmonisation of Technical Requirements forPharmaceuticals for Human Use,ICH)于2014年11月正式发布实施《Q3D:元素杂质指导原则》,并于 2022年4月更新至 Q3D(R2)[1]。ICH Q3D 指导原则依据元素的毒性及在药物中出现的可能性对元素杂质进行了分类,提供不同给药途径(口服、注射、吸人、皮肤)下元素杂质的安全性评估原则,并介绍基于风险评估原则的药物中元素杂质的评估和控制方法。根据ICHQ3D 指南,元素杂质检测方法可用于元素杂质风险评估和质量控制。早期采用的重金属检测法因专属性和灵敏度均较差、不能对单个元素杂质进行定量分析等缺点[2],已不能满足 ICH 指南对元素杂质控制的要求。随着分析技术的发展,已有多种现代仪器方法被用于元素杂质检测,如原子吸收光谱法(atomicabsorption spectrometry ,AAS)[4-5]、X荧光光谱法(Xray fluorescence ,XRF)[6-7]、电感耦合等离子体原子发射光谱法(inductively coupled plasma-atomic emissionspectromety,ICP-AES)[89]、电感耦合等离子体-质谱法(inductively coupled plasma-mass spectrometry, ICPMS)[8-10] 等,其中 ICP-AES 和 ICP-MS 是美国药典United States pharmacopoeia,USP)<233>推荐的两种检测方法,中国国家药典委员会发布的《元素杂质指导原则》(第三次公示稿)也指出ICP-AES 和 ICP-MS是同时测定多种元素杂质的常用方法。为了更好地开展元素杂质风险评估,制定适当的质量控制策略,科学合理地进行元素杂质检测方法的开发及验证至关重要。因此,笔者在本文对 ICP-AES 和 ICP-MS 的方法开发基本思路以及方法学验证要求进行了探讨,以期为相关研究人员提供参考。
1.ICP-AES 和 ICP-MS
ICP 是一种通过随时间变化的磁场电磁感应产牛电流作为能量来源的等离子体源。通过ICP 源的光谱分析等离子体原子组分的方法即是ICP-AES,而当ICP 源用作质谱分析的离子源,分析组分的方法即是ICP-MS。ICP-AES 和 ICP-MS 的共同优点是测试元素范围广,灵敏度高,可以同时快速分析多种元素,并且具有较宽的动态线性范围,是各国药典主要推荐的元素杂质检测方法。但由于检测系统的差异,ICP-MS 与ICPAES 的灵敏度存在差距,ICP-MS 的检测灵敏度更高最低可以达到 10-9水平,而 ICP-AES 仅能达到 10-6水平,故 ICP-MS 更适用于痕量和超痕量元素分析[2-3]。
尽管 ICP-AES 和 ICP-MS 有很多优点,但采用ICP-AES 和 ICP-MS 测定元素杂质会受到光谱和非光谱干扰的影响。与ICP-AES 比较,ICP-MS 受到的干扰更多。研究显示,由于空间电荷效应[11],导致 ICP-MS中存在明显的信号抑制现象,残留碳含量高的消化剂[12-13]导致某些元素分析信号增强或抑制,以及由(Ar)和碳(C)组成的多原子离子[14]影响铬(Cr)同位素的测定等多种类型的干扰问题。因此,采用ICP-AES 和 ICP-MS 分析元素杂质之前应充分了解药品的成分,识别潜在的干扰,并选择适宜的校正方式。
2.方法开发
2.1 确定待测元素种类
药品中元素杂质有多种来源,可依据 ICH Q3D 中方法全面评估元素杂质的潜在来源,主要包括原料药、辅料、生产设备、生产用水、包装材料等,每一种来源的潜在贡献都需考虑,以确定元素杂质对药品的整体贡献情况。在了解潜在来源的基础上,需结合给药途径、剂型特点、组成成分的理化特性、生产工艺等信息,确认各潜在来源中需要研究的元素杂质种类。ICH Q3D[1]中指出,针对所有给药途径,对于有意添加的 1、2A、2B 和(或)3 类元素都需要进行风险评估。针对非有意添加的元素,则需按不同给药途径的要求进行分析评估。其中,对于口服给药途径,需分析评估1类和2A 类元素;对于注射给药途径,需分析评估 1类和 2A 类元素,以及3 类元素中的锂(Li)、锑(Sb)、铜(Cu);对于吸入给药途径,需分析评估1类、2A 类元素,以及全部3类元素。此外,对于液体和半固体制剂,元素杂质从包装系统中浸出的可能性高,通常需对此来源的元素杂质进行评估。
2.2 确定各元素的限度
ICH Q3D 中提供 24 种元素杂质在不同给药途径下的每日允许暴露量(pemmmitteddaily exposure,PDE),并且为了更便于进行药品或其组分中元素杂质含量的评估,提供4种将 PDE 值转换为浓度限度的计算方式,在所获得的浓度限度能够确保药品不超过 PDE 值的前提下,研究者可选择任意方法。对于 ICH Q3D 中未列出 PDE 值的元素,可以按照 ICH Q3D 附录1中提供的方法,利用动物研究结果建立暴露限度。此外,也可以参考官方发布的其他指南确定限度,如欧洲药品评价局(European Agency for Evalution of Medical Prducts,EMEA)发布的金属催化剂或金属试剂残留限度指南(EMEA/CHMP/SWP/4446/2000)[15] 中列出铁、锰、锌的 PDE 值;美国食品药品管理局( Food and Drug Administration,FDA)发布的21 CFR 73.1200(c)[16]规定药品中铁的 PDE 值21CFR 201.323[17]规定全静脉营养液中铝的限度值。
2.3 前处理方法选择
大多数情况下,元素杂质在药品中通常以痕量或微量的浓度存在,且存在于相当复杂的基质中。因此,ICP-AES 和 ICP-MS 检测需要开发适宜的样品制备程序,以降低样品制备过程中的元素损失和二次污染风险。USP<233>规定了4种优选的前处理方法,包括直接测定法、直接溶解法(水溶液)直接溶解法(有机溶液)、间接溶解法,其中直接溶解法和间接溶解法应用最为广泛,但由于需要分析的药物种类繁多,该通则并未给出详细的样品制备方法。一般而言,分析人员应根据样品的性质选择合适的样品制备方法并对方法进行确认,而不应简单地以样品溶液是否澄清为依据。此外,在选择适当的稀释剂/溶剂来溶解样品成分或用于消解样品的试剂时,需考虑样品的化学稳定性和挥发性,以及待测元素的稳定性,避免引起目标元素的损失。表1汇总近年来一些文献报道的不同结构特征原料药以及不同类型药物制剂采用ICP-AES 和(或)ICP-MS 检测元素杂质的样品制备方法。
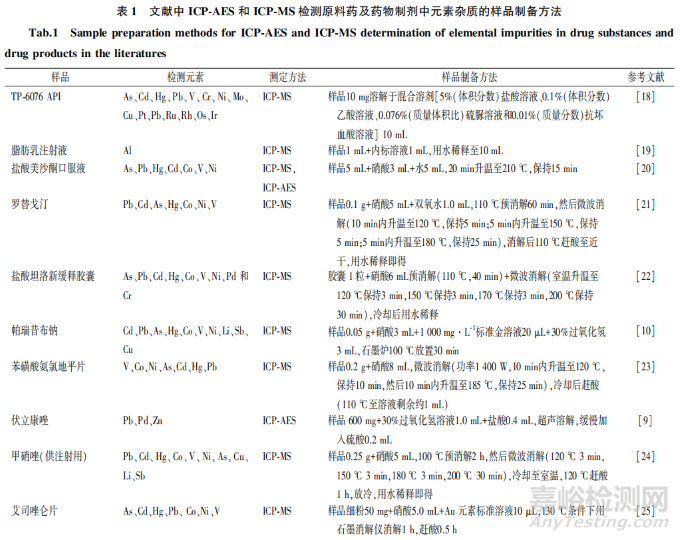

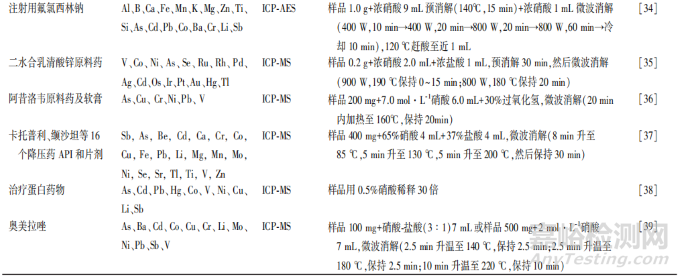
2.3.1 直接溶解法
直接溶解样品的方法简单、操作步骤少,且元素杂质损失较少,是首选的样品制备方法,尤其是对于可溶于水的样品。水溶性样品如无机盐,可采用稀硝酸直接溶解,或用浓硝酸溶解后再加水稀释以降低酸的浓度,硝酸浓度通常在 1%~10%。在一些情况下,可以加人较低浓度(0.1%~1%)的盐酸比如在检测汞( Hg)(Ⅱ)[40]、钯(Pd)(Ⅱ)[41]或铂(Pt)(W)[42]的离子时,盐酸可作为一种络合剂稳定上述离子,避免在样品引人系统的延迟及任何记忆效应,或者在锇(0s)检测中加人盐酸保持其稳定性!。汞元素的检测中,金(Au)(Ⅲ)离子也可以起到类似盐酸的作用,使汞元素在样品中保持稳定,并降低测量过程中的记忆效应[43]。
对于不溶于水的样品,可用有机溶剂进行溶解,其检测仪器需要采用膜去溶剂化装置或者雾化器通入氧气[8] 。常用的有机溶剂包括 N,N-二甲基甲酰胺[44]二乙二醇单乙醚[45] 、2-丁氧基乙醇[46]、乙醇[47]等,但是对于 ICP-AES 和 ICP-MS 而言,过多有机物进人系统内易导致雾化器和等离子矩管积碳、积盐,进而影响仪器灵敏度,因此需加人水尽可能地降低有机溶剂的浓度。
2.3.2 间接溶解法
不能直接溶于水或有机溶剂的样品,尤其对于以不同成分(原料药、包衣、填充剂、粘合剂)以及不同剂型(片剂、胶剂、凝胶剂、软音剂)存在的药物制剂,通常需采用消解方法消解样品,从而使其完全溶解,同时需注意最终样品溶液中的残碳量要相对较低,以降低测量过程中出现干扰的可能性[48]。间接溶解法包括敞开式消解法和密闭消解法,敞开式消解法易导致挥发性元素的损失,同时可能引入外部环境的污染,因此一般推荐采用密闭消解法制备样品,可最大限度降低挥发性元素的损失。
酸的选择应基于样品的基质,优先选择硝酸消解,也可以采用盐酸、硫酸、氢氟酸,或硝酸与其他酸的混合酸(如王水[盐酸:硝酸=3:1])。当样品中含有氧化硅、二氧化钛或滑石粉时,可以加入氢氟酸将其完全溶解[49],不过需确认仪器是否适用于氢氟酸进样,对于一些易挥发性元素,可添加稳定剂保证结果准确,如汞(Hg)元素的检测中加人0.5%~5%盐酸或 Au3+离子[50],锇(0s)元素检测中加人含有 0.01 mol·L-1硫脲及0.1g·L-1维生℃的 0.5% 乙酸[51]、含有0.009 mmol·L-1KBr0;的 1% 盐酸溶液]或0.01 mol·L-1硫脲[40]。在加压密闭容器中进行微波辅助湿法消解也可以减少元素的损失,同时还可以提高方法的准确度和重复性[36],目前已成为难溶性药物的常见样品制备方法[21-24,26-28,30,32,36-37,39]。此外,需要注意的是,若消解后供试品溶液酸度较大,则需要赶酸以保证供试品溶液酸度与标准溶液一致,同时消解后赶酸也可以起到保护仪器的作用。
2.4 千扰和校正
ICP-AES 在测量过程中可能会由于分析谱线的重叠而产生光谱干扰,为了追踪这些干扰并选择无干扰的分析谱线,可以将特定元素的测量结果与使用其他替代谱线得出的结果进行比较[50]。STOVING 等[43]在对 LU(代号)药片 18 种元素杂质检测研究中,每种元素都选择3种分析谱线进行测定,并通过加标回收率的相对标准偏差(RSD)验证结果以及目测评估,最终除铱(Ir)、Os、Pb、Pt 和铑(Rh)外,其他元素都选择出发射强度高且无光谱干扰的分析谱线。
ICP-MS 同样会受到光谱重叠的干扰,其中最常见的类型为多原子离子干扰,通常与所使用的等离子体/雾化器气体、溶剂/样品中的基质成分、样品中的其他元素或周围空气中夹带的氧/氨有关[52] 。现代 ICP-MS仪主要通过碰撞池和/或反应池来消除或减少来自于等离子体和基质的多原子离子干扰[53],目前常见的碰撞气体有氦(He)和氙(Xe),反应气体有氧气(02)、氨气( NH3)、甲烷(CH4)和氢气(H2)等。已有文献报道使用 NH3的动态反应池测定原料药、口服片剂、肠外溶液中的元素杂质时,避免铬、铜、锰、钒的多原子干扰[54];使用 He 的碰撞池测定肝素钠中的元素杂质时避免K、Cr、V、Fe、As 和 S的质谱干扰[55]。另外一种消除光谱重叠的有效方法是使用基于不同质量分析器(扇形磁场、飞行时间和离子阱等)的高分辨质谱仪,其通过较高的分辨能力来有效解决多原子离子和同位素干扰问题,但由于操作技术的复杂性、以及仪器自身和运行维护的成本较高,只有少数专业实验室在使用[56]同质异位素干扰也是 ICP-MS 中一种常见的质谱干扰除上述解决方式外,在一些情况下也可通过选择合适的同位素来消除此类干扰。王妮等[57]通过为 Cd、Sb、Pb选择合适的测试同位素,成功地分析3者在氟比芬酯脂微球注射液玻璃包装中的迁移量。此外,在等离子体中引人混合气体(如 Ar 与 N2、02的混合气体)[45],以及一些新的样品引人系统,如冷蒸汽发生、氢化物发生、膜脱溶,冷等离子体技术等[54,58] 也已有报道用于解决光谱干扰。
对于 ICP-AES 和 ICP-MS 测量中存在的非光谱基质干扰,当元素检测灵敏度可以保证时,降低基质效应最简单的方法就是样品稀释,或者也可以选择合适的样品引入系统,如使用V形槽雾化器来分析总溶解体量含量较高的样品溶液。标准加人法则是通过在供试品溶液中加入不同浓度的待测元素标准溶液,来校正样品的基质效应。谢莉等[9]采用标准加人法-ICP-MS 测定了氢化可的松注射液中的21 种元素杂质,各元素回收率均在 90%~110%,方法准确度较好。引人内标元素也是校正基质效应的一种常用方法,内标的作用是补偿不同样品之间基质成分的差异,校正与样品溶液气动雾化有关的物理干扰以及可能受基质成分影响的传输过程。在使用 ICP-AES 时,通常对所有测量元素使用一种内标,如钪(Se)或钇(Y)。但对于ICP-MS,由于所测定元素信号的抑制或增强幅度与质量有关,一般需要多个内标才能覆盖整个质量标样,因此通常为低、中、高质量数元素选择内标 3 或4个[50]刘宇澄等[28]采用 ICP-MS 分析兰索拉唑原料药中元素杂质,测定 Li、Co、V、Ni 时以45Se 为内标,测定 Cd、Sh时以103Rh 为内标,测定 Pb、Hg 以193Ir 为内标。需注意的是样品中应不含有内标元素,且内标元素与待测元素的质量数应相接近。另外,通过调节仪器参数(如样品提升速率、雾化气流速、射频功率、采样深度和离子透镜电压等)虽然不能消除基质效应,但是在一定程度上可以降低基质效应的影响程度。
3.分析方法验证
药物分析方法验证实施通常主要参考ICH Q2、USP <1225>以及《中国药典》通则 9101。2023年 11月发布的 ICH 02(R2)扩大适用的检测方法类型,并且为了适应更多类型的方法,对于方法学验证的要求也进行了较大的修订。ICH Q2(R2)现已适用于ICP-AES和 ICP-MS 方法,同时为了更好地指导分析人员运用该指南理念进行方法验证,在指南附件2中也提供ICP-AES 或 ICP-MS 测定元素杂质的验证方式示例,但需说明的是该示例并非强制性要求。除 ICH Q2(R2)之外各国药典通则,包括 USP <233>、USP <730 >、欧洲药典(European phammacopoeia,EP)2.4.20 EP 2.2.58.和《中国药典》通则 0412 也都有方法学验证的相关内容。此外,我国药典委员会发布的《元素杂质指导原则》(第三次公示稿)(下文统称药典委员会公示稿)中也介绍元素杂质方法学验证的一般要求。表2汇总对比上述指南中关于元素杂质测定方法的方法学验证要求。
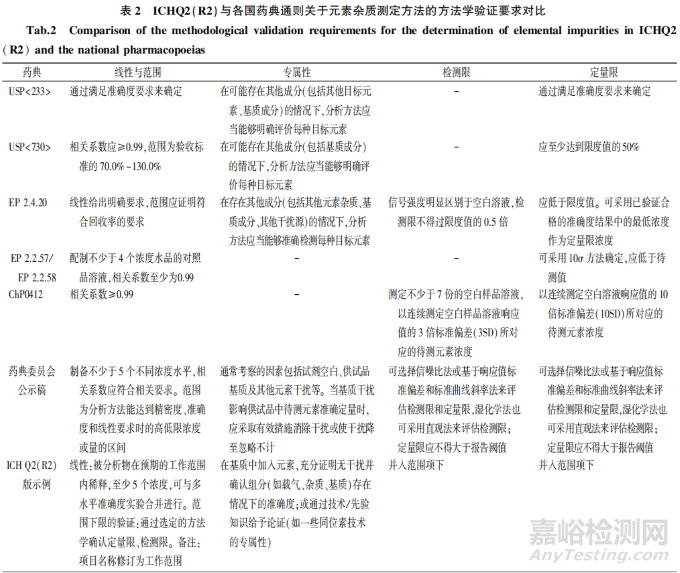

3.1 专属性
采用ICP-AES 和(或)ICP-MS 法进行元素杂质检测时,可能存在不同来源的干扰,需对方法的专属性予以验证。USP 和EP 相关通则中均明确要求在其他成分(如其他元素杂质、基质成分、其他干扰源)存在的情况下,分析方法应当能够准确检测每种目标元素。ICH Q2(R2)和药典委员会公示稿的示例中提供需要考察的干扰源包括空白试剂、供试品基质、载气、其他元素和杂质,药典委员会公示稿更进一步强调若基质存在干扰,应采取有效措施消除干扰或使干扰降至忽略不计。
3.2 范围
ICH Q2(R2)版中,将线性、范围下限(定量限、检测限)统一并人范围项下,强化了范围的重要性,对于 ICP -AES 和 ICP-MS 方法,被测元素浓度与响应之间呈线性关系,ICH Q2(R2)示例中要求线性验证至少5个浓度,这与药典委员会公示稿要求相一致,并且ICH Q2(R2)正文规定对于线性响应需提供数据图、相关系数或决定系数、Y 轴截距和回归线的斜率。但是,ICH Q2(R2)正文及示例部分、药典委员会公示稿均未对相关系数做出规定,USP、EP、ChP 相关通则均明确要求相关系数不低于0.99。此外,鉴于药品中的元素杂质通常以较低水平存在,因此方法的灵敏度评价非常重要。各通则/指南对于范围下限的验证均包括定量限、检测限,其中 ICH Q2(R2)和药典委员会公示稿提出的验证方法最为全面,包括直观评估法、信噪比法基于响应值标准偏差和标准曲线斜率法,不过药典委员会公示稿未给出各方法的具体要求,ICH Q2(R2)正文中则明确采用信噪比法时,一般可接受的检测限信噪比为 3:1,定量限信噪比至少为 10:1;采用基于响应值标准偏差和标准曲线斜率法时,检测限为3.3σ/S定量限为 10σ/S(σ =响应值的标准偏差;S=标准曲线的斜率)。对于范围下限与元素限度值(PDE)之间的关系,ICH Q2(R2)未予以规定,仅明确对于杂质检查,分析方法的定量限应等于或低于报告阈值,而EP24.20 明确规定检测限不得过限度值的0.5倍,定量限应低于限度值:USP<730>对定量限的要求则更为严格需至少达到50%限度值。另外需要提醒的是,ICH Q3D中规定元素 PDE 的 30%为控制阈值,其作为是否需对药品元素杂质进行额外控制的判断指标,从这个角度而言,如需使用控制阈值时,元素杂质分析方法的定量限则不能超过 PDE 的 30%。
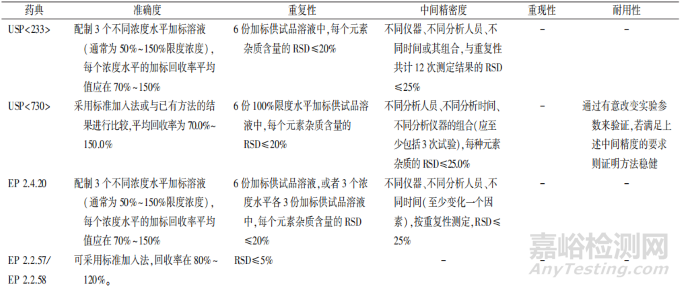
3.3 准确度
ICH Q2(R2)版示例对于方法的准确度验证仅简单说明了采用杂质的加标研究或与正交方法对比杂质谱,但在其正文中举例可以在3个浓度各重复测定3次进行验证,并且说明在某些情况下,如果精密度、范围内响应和专属性已经确证,方法准确度可以通过推论得出。USP、EP 及药典委员会公示稿对于确度的验证要求则相对具体很多且基本一致,即供试品加标溶液配制3个不同浓度,添加水平通常在50%~150%限度浓度范围内,各元素每个浓度的平均回收率应在 70%~150%范围内。在上述要求基础上,药典委员会公示稿对于加标浓度还予以更为细致的说明,包括加标溶液为高、中、低3个浓度水平且每个浓度至少制备3份样品,并且指出当供试品中元素杂质含量较低时,可设计更低的浓度点如报告阈值水平。
3.4 精密度
方法精密度验证包括重复性、中间精密度、重现性3个方面。①重复性:ICH Q2(R2)示例提供两种验证方式,一种方式是在可报告范围内取3水平各重复测定3次,另一种方式是 100%试验浓度测定6 次。对于第一种方式,可以引用准确度的验证结果这也符合 ICH Q2(R2)正文中对于准确度和精密度可以合并评价的建议。EP 2.4.20、药典委员会公示稿同样提出了上述两种验证方式,并进一步要求每个元素茶质测定结果的 RSD≤20%。②中间精密度:USP、EP通则及药典委员会公示稿对于中间精密度的要求基本一致,变动因素均包括不同时间、不同分析人员、不同仪器,ICH Q2(R2)版则在上述因素基础上增加了不同环境条件的要求。中间精密度的具体可接受标准在ICH Q2(R2)正文及示例中均未给出具体规定,但USP、EP 和药典委员会公示稿均明确要求测定结果的RSD≤25%。③重现性:重现性在 USP、EP 通则中均末提及,但药典委员会公示稿说明在方法转移或方法复核时,应考察方法的重现性。ICH Q2(R2)则是在正文中说明递交申请时通常不需要考察重现性吗,但当分析方法需要收载到药典中以及在多个场地使用时,则应考虑予以研究。
3.5 耐用性
对于方法耐用性验证,药典委员会公示稿和 ICH Q2(R2)示例中给出了一些可微调的参数,包括样品消解程序、样品制备中所用关键试剂( 如酸、碱)的量或浓度、雾化器和鞘流设置、等离子体设置。药典委员会公示稿在耐用性项下还提出了供试品溶液的稳定性验证。
总之,在良好方法开发的基础上,ICP-AES 和 ICPMS 用于药物元素杂质检测还需通过全面的方法学验证,分析人员可以依据 ICH Q2(R2)及各国药典相关通则证实方法的科学性及适用性。
4.结论与展望
自 2014 年 ICH Q3D 发布,经过 10 年的发展,基于风险评估对元素杂质控制的理念,有效促进了药品整体质量水平的提高。ICP-AES 及 ICP-MS 方法以灵敏度高和专属性强等优点,如今已广泛用于药物中元素杂质的检测并进行风险评估和质量控制。本文综述了这两种方法在元素杂质检测方面的方法开发要点和具体的方法学验证原则,可提高分析方法开发的效率,有助于保证分析结果的准确性和可靠性。值得重点关注的是,目前药品前处理方法还是需要具体问题具体分析,由于药品的基体具有类似性,因此对前处理方法规律进行归纳总结,将会对方法开发具有指导意义:另外,目前 ICP-AES 和 ICP-MS 前处理较为复杂且耗时对于难溶性原料药、药用辅料和固体制剂通常需加入浓酸或有机溶剂,因此发展灵敏度高、快速无损、绿色环保的分析技术(如x射线荧光光谱法、激光烧蚀-ICP-MS 等)将会是一大重点研究方向,同时有助于实现药物元素杂质的过程控制和/或现场检测。

来源:医药导报


