您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-09-15 06:53
近几年随着国内医疗器械行业在高科技、新技术等方面突飞猛进地发展,涉足研发高科技、高风险医疗器械的国内企业也逐渐多了起来。海外市场的布局也成为这些企业规划的方向和目标。美国作为海外市场的主要国家,拥有全球最好发展医疗器械的平台和环境,取得高风险医疗器械在美国上市许可也成为这些企业目标。
下面我们就来介绍一下美国高风险的医疗器械(III类)上市途径“上市前批准Premarket Approval (PMA) ”。
上市前批准(PMA)概述
上市前批准(PMA)是FDA对III类医疗器械的安全性和有效性进行科学和监管审查的过程。PMA是FDA要求的最严格的器械上市申请类型。在器械上市之前,申请人必须获得FDA对其PMA申请的批准。PMA批准是基于FDA的一项决定,即PMA包含充分有效的科学证据,以确保器械对其预期用途是安全有效的。PMA申请人通常是发明者或开发者和最终的制造商。
有关上市前批准(PMA)的法规位于《联邦法规》第21篇(CFR)第814部分,医疗器械上市前批准。
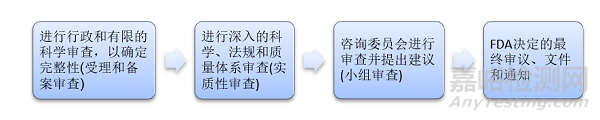
上市前批准(PMA)评审流程
上市前批准 (PMA)的评审是一个四步骤的审查过程,包括:
上市前批准PMA信息要求
上市前批准(PMA)申请是向FDA证明III类器械安全性和有效性的科学、规范的文件。PMA申请有行政要素,但良好的科学和科学写作是PMA申请获得批准的关键。如果PMA申请缺乏行政清单中列出的元素,FDA将拒绝PMA申请并将不进行科学和临床数据的深入审查。如果PMA申请缺乏有效的临床信息和合理的科学推理的科学分析,它可能会影响FDA的审查和批准。PMA申请不完整、不准确、不一致、缺失关键信息和组织不好将导致这些申请的延迟批准或拒绝。制造商应在将PMA申请提交给FDA之前进行质量控制审核,以确保其科学合理,并以组织良好的格式呈现。
上市前批准(PMA)质量体系
设计控制
必须建立和维护程序来控制器械的设计,以确保满足规定的设计要求。设计控制包括建立和维护描述设计和开发活动的计划,并定义实施的责任。计划必须识别并描述为设计和开发过程提供或导致输入的不同组或活动之间的接口。设计过程还包括:
√进行风险分析;
√确定器械的设计输入或要求;
√制定器械的设计输出或规格;
√验证设计输出满足设计输入;
√在设计过程中适当的时间进行设计评审,以确定设计或设计过程中的重大问题;
√确认设计满足制定的用户需求和预期用途;
√验证器械中使用的任何软件;
√将器械设计转化为生产规范;
√在设计过程中控制设计的变化和上市产品设计的变化;
√在设计历史文件中记录设计控制活动。
PMA提交文件应包括制造商实施以符合QS法规(21 CFR 820质量体系法规)的设计控制的完整描述。设计历史文件需要提供给FDA检查。如果缺乏这些信息,FDA将无法完成上市前审查过程。
生产控制
PMA提交必须包括器械的制造、加工、包装、存储以及器械安装使用的方法、设施和控制的完整描述。描述必须包含足够的细节,使熟悉质量体系/良好生产规范要求的人能够对器械制造中使用的质量控制做出判断。
器械制造商不仅包括制造或组装成品器械的公司,还包括PMA申请人运营或承包的公司,以执行生产操作的一部分,如灭菌或包装。合同制造商可以直接向PMA申请人提供适用于其操作的所需生产信息,以纳入PMA提交,或直接在器械主文件中向FDA提交此类信息。PMA申请人应提供在PMA提交中引用器械主文件信息的书面授权。
质量体系检查
产品评估和质量办公室(OPEQ)审查PMA提交的质量体系设计和制造信息。产品评估和质量办公室(OPEQ)确定制造商是否对工艺进行了足够详细的描述,并初步确定制造商是否满足质量体系要求。如果制造商提供了足够的设计和制造过程的描述,可以开始预批准检查。
当产品评估和质量办公室(OPEQ)确定制造商已在PMA提交文件中证明其设计和制造工艺符合QS法规(21 CFR 820质量体系法规)要求,且工厂已准备好接受检查时,将发出检查任务。
检查将包括评估公司在PMA提交文件中声称的设计和制造器械的能力,并确认公司的质量体系符合21 CFR 820质量体系法规。检查过程考查公司建立正式QS程序的程度,并确保通过工艺验证批准的设计被恰当地转化为规范。检查级别为FDA现场检查2级全面检查,2级检查将覆盖所有四个主要子系统(管理控制、设计控制、纠正措施(CAPA)和生产控制)。

来源:海河生物


