随着科技的进步,新的技术、设备、新的科技成果越来越多的应用在药品研究生产领域,对药品研发和已上市药品的质量提升起到了重要作用,由此带来的药品生产过程中的变更是生产常态,也是客观必然。生物药研发过程中是允许对工艺过程或参数等进行优化的,这种优化不只存在于临床开发阶段,在药物上市后也是会经常发生的。但是对于这种变化所引入的安全性和有效性风险需要谨慎评估。无论蛋白药物还是细胞治疗,药学变更后的可比性研究都不是一项简单的任务。可比性研究的核心目的是确认变更前的非临床和临床数据适用于变更后的产品。本文主要就非临床和临床桥接研究进行概述(药学可比性研究不在本文讨论之列)。
生产工艺改变是不可避免的
通常来讲,生产工艺变更越早其实越好,最好出现的是低风险变更。但实际情况是,工艺变更既有低风险也有高风险,变更发生在药物开发过程中,也发生在药物上市以后(Process changes happened anytime and anywhere)。分享几个变更的案例可能更容易理解一些。
1)重组蛋白:Shingrix(Herpes Zoster Vaccine)
本品主要发生的药学变更内容包括:生产规模扩大、表达工艺优化使抗原表达量提高、纯化工艺优化使抗原收率增加、增加pH处理步骤确保病毒充分清除、增加原液-45℃长期稳定性研究、生产场地变更。关键Ⅲ期临床研究采用工艺变更后样品开展。
2)抗体:Ilumetri(Tildrakizumab)
本品临床研究期间,平行开发了商业批原液生产工艺。工艺一的原液用于非临床研究。工艺一经生产规模放大后转移到新的设施生产。新生产的产品用于临床Ⅰ、Ⅱ期研究。目前还未涉及到工艺改变,依然使用的工艺一。不过,后续对工艺一进行了改良,形成了工艺二。工艺二较工艺一的区别主要包括:WCB代替了MCB、column changes、制剂处方变更等。关键Ⅲ期临床研究采用工艺二生产的样品开展。
3)单抗:Bavencia(Avelumab)
这个产品主要经历了两套生产工艺。工艺A的样品用于非临床和早期临床研究。工艺A优化后形成工艺B。最初Avelumab的制剂是10 mg/mL,灌装8mL,用于非临床研究、临床Ⅰ/Ⅱ期研究。为了支持产品商业化,开发了更高浓度20 mg/mL的制剂,该制剂通过工艺B生产的原液所得,并用于后续所有Ⅲ期临床研究,与拟上市制剂是一致的。
以上列举的是临床研究阶段药学工艺的变更。上市后药学变更也是很常见的。据统计,2014年10月以后EMA上市的29个单抗,申请了超过400个上市后生产工艺变更。以药王Humira为例,自2003年EMA批准上市以来,前后经历了20次生产工艺变更,包括扩大生产规模、增加新的生产设施、improvement in process controls and robustness等。
药学变更随之出现的问题就是变更前后的可比性研究。针对这一问题的指南包括ICH Q5E,Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process。
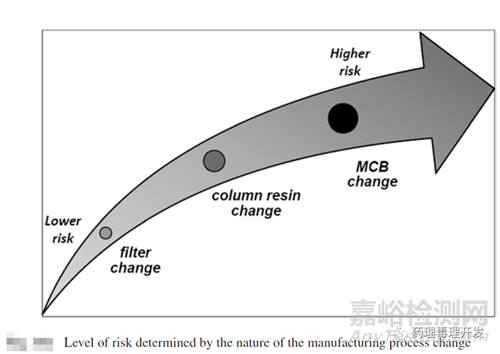
对于工艺变更可比性研究,最先需要做的是判定工艺变更的风险等级(Level of risk by type of process change)。风险等级大小直接决定了需要开展的实验或验证的数量。低风险等级的变更,有的可能只需要书面说明合理性,并不需要开展具体实验研究。高风险等级可能意味着需要开展大量产品表征、稳定性研究,甚至可能的临床研究。下图是个风险等级的简单示意图。
风险等级的划分有时是有主观成分在内的。因此,不同监管机构EMA、FDA、WHO对风险等级进行了约定。对风险的称谓略有不同,但代表的意义类似。比如对于重大风险变更,FDA称为prior approval supplement,即需要监管机构批准后方能执行,EMA则称为TypeⅡ变更,按数字进行级别划分,WHO与FDA则类似。对于中等风险变更,FDA称为changes being effected in 30days,即需要等监管机构30天进行批复,EMA则称为TypeⅠB变更。对于低风险等级变更,FDA称为annual report,即年度备案,EMA则称为TypeⅠA变更。具体哪些改变属于重大风险等级,内容较多,可自行翻阅EMA、FDA、WHO相关指南。我国CDE依据风险和产生影响的程度由高到低分为:重大变更、中等变更、微小变更三大类。
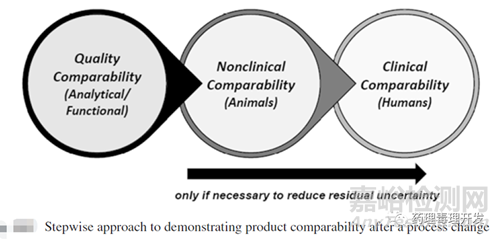
可比性研究通常是分级逐步开展。首先是质量对比,如果依然存有担忧,则进行非临床对比,如果还不能解决,则进行临床研究,如下图所示。
可比性研究受研发进程、分析方法适用性、对工艺和药物的认知程度等影响。早期临床试验阶段,可比性研究通常不如上市后的广泛。因此药学变更可分为临床试验期间变更和上市后变更两部分。
临床试验期间药学变更
某些可能对临床试验产生重大影响的变更,仅药学研究可能不足以评估变更带来的影响,还需要考虑开展非临床研究和/或临床桥接研究,如新主种子批变更、特殊辅料变更、延长减毒活疫苗生产用毒种代次、重新亚克隆筛选等。《临床试验期间生物制品药学研究和变更技术指导原则》对临床试验期间,对安全性有潜在重大影响的生物制品药学变更事项有详细介绍,可自行翻阅。
另外,如果可比性研究结果显示药学变更对临床试验的安全性或有效性可能产生负面影响(如改变免疫原性、产生新杂质等),或当仅用药学分析数据无法排除变更可能对有效性和安全性的负面影响时,可能需要包括PK/PD和毒理数据在内的非临床研究,甚至需要进行变更前后的临床桥接研究(参见:临床试验期间生物制品药学研究和变更技术指导原则)。
已上市生物制品药学变更
《已上市生物制品药学变更研究技术指导原则》中指出,若变更前后产品的生产工艺、质量和稳定性研究足以证明可比,则无需对变更后产品实施非临床和/或临床研究。但当特定质量属性与安全性和有效性之间的关系尚未确定,且观察到变更前后产品的质量属性存在差异的情况下,应实施非临床和/或临床桥接性或确证性研究。非临床和/或临床研究的方式和程度应结合药学可比性的结果、对产品性质了解的知识水平、已完成的相关非临床和/或临床研究数据以及该药物的用途,基于具体问题具体分析的原则来确定。鼓励通过药学与非临床的方式开展变更研究,若在此基础上仍无法证明可比性,应进一步考虑进行临床研究。
某些对生物制品可能产生重大影响的变更,如新主种子批重大变更、特殊制剂的关键辅料变更等,应考虑开展非临床和/或临床桥接研究。
《已上市生物制品药学变更研究技术指导原则》第五部分对生物制品常见变更类型及技术要求有详细列表说明,针对哪些变更需要非临床和/或临床数据支持也有介绍,内容较多,可自行对号入座,不再赘述。简单讲,大部分重大变更如表达载体变更、主种子批变更、主细胞库变更、工作种子批变更、培养基成分变更、动物源/非动物源材料变更、变更生产厂房/厂房/生产线、替换工艺过程控制参数和范围限度、删除工艺过程控制参数和范围限度、放宽范围限度、制剂浓度变更、制剂体积变更、变更制剂处方中辅料组成或浓度、替换原有辅料、去除辅料、佐剂浓度变化等,应考虑进一步开展非临床和/或临床的桥接研究。当然,有些变更还要甄别药学可比性研究数据是否不足以支持变更可比性。
但其实现有指南还是有些局限性,只针对哪些变更需要非临床或临床数据支持,但对于具体需要哪些非临床试验或临床试验是没有约定的,只是提示根据药学变更前后差异,决定非临床或临床试验的数量,是只开展PK研究,还是PK/PD研究,或是毒理研究,并未做太多说明。ICHQ5E的2.5部分有简单介绍非临床和临床的考虑,但也比较笼统。CDE药理毒理学部孙涛老师在2014年一篇文章《桥接研究在药物非临床研究与评价中的应用》中提到,针对不同的情况,桥接研究的思路和重点有一定差异,但通常是以药动学比较研究为先导和主线的。所以,PK研究往往是要做的。但由于生物制品药学变更复杂多样,CDE鼓励按照《药品上市后变更管理办法(试行)》相关要求,通过沟通交流途径,就预期的变更类别、可比性方案和研究内容等与监管机构进行沟通,特别是对于拟进行重大变更的产品。
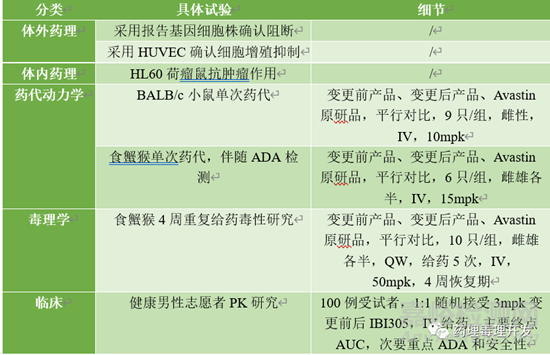
最后分享一个最近信达生物刚发表的《Comparability strategy and demonstration for post-approval production cell line change of a bevacizumab biosimilar IBI305》,属于已上市生物制品变更细胞株。当然,这肯定属于重大变更了。其开展的非临床和临床研究如下。
非临床桥接的内容与药学工艺变更的阶段、变更的大小密切相关,但药学工艺变更通常又是无法避免的。先确定变更阶段,是临床阶段还是已上市阶段。再确定变更级别,是重大变更、中等变更、还是微小变更,结合FDA、ICH、EMA、NMPA的具体指导原则,对号入座,确定变更对非临床及临床研究的影响,再制定具体的桥接研究计划。当然,指南通常只提供大的框架和思路,详细研究计划可以跟监管机构或业内同行、专家讨论、沟通。