三年的疫情给医疗器械行业带来了诸多“利好”。期间,很多企业赚的盆满钵满,着实让其他行业羡慕不已。
再加上近年来,“医疗器械注册人制度”全面实施,使得“两证”得以捆绑,大幅度降低了厂房、设备等投资成本。
于是,新年伊始,很多企业蓄势待发,准备扩大规模;还有很多新进行业的企业,准备浩浩荡荡地大干一场。
但是医疗行业,严重受制于法律法规,这一点不同于其它制造业。
为了避坑,让大家少走弯路、少交学费,甚至不要以身试法,本篇分享医疗器械从研发到上市需要经历的九大过程。
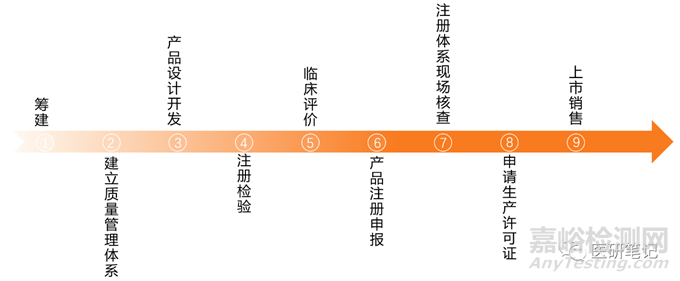
No.1 筹建基础设施
1) 注册公司,本工作比较容易,在此忽略。
2) 筹建厂房、净化车间、生产设备、检验设备等硬件。
提醒:可采用注册人制度,委托成熟的医疗器械生产企业研发或生产,则可以节省大量的硬件投资。
No.2 建立质量管理体系
企业负责人、管理者代表(高管可兼职)、质量负责人、研发负责人等均已到位后,根据《医疗器械生产质量管理规范》和GB/T 42061-2022 (ISO 13485)建立质量管理体系。
如:质量手册、文档管理控制程序、设计开发控制程序、采购控制程序、风险管理控制程序、不良事件管理控制程序、召回管理控制程序等。
注意:本过程可以与设计开发同时进行,但是时间逻辑顺序要注意。
No.3 产品设计开发
根据ISO13485,医疗器械的设计开发大致可分7个阶段:
项目策划阶段、设计输入阶段、设计输出阶段、设计验证阶段、设计确认阶段、设计输出阶段、设计转换阶段。
本阶段为项目核心的核心,项目的80%资源都集中在这个阶段。
No.4 注册检验
完成了注册检验样品后,根据《医疗器械注册与备案管理办法》,应当按照产品技术要求进行检验。
检验合格的,方可开展临床试验或者申请注册、进行备案。
并且,检验样品应具有典型性,应当能够代表安全性和有效性,并且其生产应当符合《医疗器械生产质量管理规范》的相关要求。
No.5 临床评价
根据《医疗器械临床评价技术指导原则》,临床评价分为三种途径:
1) 对列入《免于进行临床试验的医疗器械目录》中的产品,可免于临床试验。
2) 对于同品种医疗器械临床试验或临床使用获得的数据进行分析评价。
3) 按照《医疗器械临床试验质量管理规范》开展临床试验。
提醒:若开展临床试验,建议委托第三方CRO公司。
No.6 产品注册申报
该过程相当于根据药监局的要求,进行正式考试答卷。
注册资料撰写,是个庞大系统的工作过程,需要公司所有部门的配合。
1) 申报部门:
境内一类医疗器械:向设区的市级负责药品监督管理的部门提交备案资料。
境内二类医疗器械:向省、自治区、直辖市药品监督管理部门提交注册资料。
境内三类医疗器械:向国家药品监督管理局提交注册资料。
进口一类医疗器械:向国家药品监督管理局提交备案资料。
进口二、三类医疗:向国家药品监督管理局提交注册资料。
2) 按照eRPS系统要求,注册资料应包括:
以三类医疗器械为例,其他类似。
第1章——地区性管理信息
第2章——申报产品综述资料
第3章——非临床研究资料
第4章——临床研究资料
第5章——说明书、标签
第6A章——质量管理体系程序
第6B章——申报器械的质量管理体系信息
No.7 注册体系现场核查
申请人应当在申请注册时提交与产品研制、生产有关的质量管理体系相关资料。
受理部门认为有必要现场进行核查的,应当组织开展质量管理体系核查。
1) 境内三类医疗器械质量管理体系核查,由国家局器械审评中心通知申请人所在地的省、自治区、直辖市药品监督管理部门开展。
2) 境内二类医疗器械质量管理体系核查,由申请人所在地的省、自治区、直辖市药品监督管理部门组织开展。
No.8 申请生产许可证
获得注册证后,即可以向省、自治区、直辖市的药监管理部门申请生产许证。
申请时,需要提交相应的证明资料以及所生产医疗器械的注册证。
从事一类医疗器械生产的,向地级市的药监管理部门备案。
No.9 上市销售
恭喜了。
获得注册证和生产许可证后,即可上市销售了。
以上是对医疗器械从研发到上市的九大历程简析,希望对大家有所帮助。