导语
作为发达国家中的一员,韩国的医疗器械市场规模是不可小觑的,然而与美国等其他发达国家相比,目前韩国医疗器械产业的国际竞争力仍处于较低水平,相关数据显示,其国内半数以上的获准医疗器械都源于韩国境外的国家。那么本期文章我们就来跟大家分享一下韩国医疗器械注册的相关知识。
1、分类规则
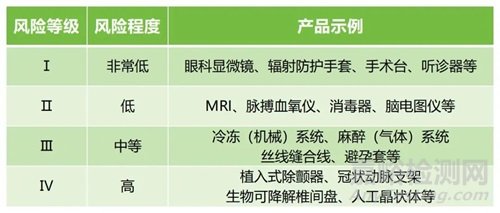
韩国根据产品对人体的潜在危害:1)与人体接触的持续时间;2)侵入性程度;3)是否向患者提供药品或能量;4)是否对患者有生物学影响,将医疗器械分为Ⅰ、Ⅱ、Ⅲ、Ⅳ四个风险等级。
可通过如下方法确定产品分类:
1. 查阅Regulation on Medical Device Groups and Class by Group的Attachment,法规中规定了分类规则以及产品的代码和风险等级,可以根据产品定义/用途等判断产品的等级,我们随附了示例图,红框的数字为产品的风险等级。
2. 在韩国已注册产品的数据库(可联系我们获取链接)中,用产品名称(英文/韩文)进行检索,查找类似产品在MFDS中的分类。
2、上市前递交路径
在韩国,Ⅰ类产品一般只需在系统上做简单的Notification登记即可,有SE Device(即实质等同产品)的Ⅱ类产品,需递交技术资料进行NIDS认证;而对于NSE Device(即没有实质等同产品)的Ⅱ类产品以及Ⅲ、Ⅳ类产品,则需要由MFDS批准。不同类别产品所需提交的注册资料不同,具体可联系我们咨询。
3、韩国代理人
和大部分国家或地区一样,在韩国没有实体办公场所的公司必须任命一名韩国许可持有人(Korean License Holder)来协调他们向MFDS注册医疗器械的事务。KLH控制医疗器械注册并帮助韩国境外制造商遵守韩国良好生产规范(KGMP)要求,并且其名字会出现在制造商的MFDS医疗器械注册证书(产品审批证书)上。产品获批后,KLH需负责进口医疗器械的年度报告事宜,并且接受MFDS的突击审核。
一般情况下,企业会选择在韩国的分销商作为证书持证人,但选择没有销售合作并有能力协助产品注册的韩国企业作为持证人,在后续需要更换经销商时可能会更加便捷。
4、质量体系要求
在韩国,Ⅱ、Ⅲ、Ⅳ类医疗器械的制造商都需要符合KGMP(Korean Good Manufacturing Practice)的要求,KGMP要求与ISO 13485类似。
KGMP证书是颁发给进口商(Importor)而不是制造商(Manufacturer), 证书每3年需更新一次,企业要在在证书过期前的90天更新。
通常对于高风险的Ⅲ、Ⅳ类医疗器械,由监管当局MFDS审核制造商的质量管理体系,而Ⅱ类和部分Ⅲ、Ⅳ类的医疗器械交由第三方机构审核。
审核现场的语言要求是韩语,因此企业需要提前准备韩语翻译。审核周期大约在9-12个月。