您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-06-24 20:20
摘 要: 建立一种检测降血压、降血脂和降血糖类保健食品中58种非法添加药物的高效液相色谱-高分辨质谱分析方法。样品经酸化乙腈提取,采用基于QuEChERS原则的快速净化技术,以0.1%甲酸-5 mmol/L甲酸铵水溶液和甲醇-乙腈(体积比为1∶1)为流动相,进行梯度洗脱,经ESI正模式扫描,建立了包括58种药物的结构信息、一级/二级精确质量数等在内的数据谱库,用于未知样品二级质谱图自动匹配确证。保健食品中58种非法添加物可同时进行定性及定量检测,线性关系良好,相关系数不小于0.999 0,检出限为0.01~10 ng/mL,定量限为0.03~30 ng/mL。平均回收率为64.25%~115.3%,相对标准偏差为1.1%~17.85%(n=6)。该方法样品处理简单,灵敏度高,特异性强,可为保健品中降三高类非法添加物质的高通量筛查确证提供方法参考。
关键词: 高效液相色谱-高分辨质谱法; 降血压; 降血脂; 降血糖; 非法添加; 保健食品
高血压、高血糖和血脂异常是慢性心血管疾病(CVD)的主要危险因素[1‒2],我国面临人口老龄化问题,CVD的发病率逐渐升高,降三高成为我国公共卫生政策的重点[3‒4]。高血压及代谢异常通常会合并存在,比如高血压患者合并血脂异常、胰岛素抵抗、肥胖等,这些疾病独立或聚集可导致脑卒中等较为严重的心血管疾病[5‒6]。心血管疾病的治疗需要长期稳定服用药物甚至需要联合用药[7‒8],许多患者对此具有抵触心理,加之保健食品行业日渐兴起,越来越多的人更倾向于所谓的无副作用辅助降三高的保健食品。然而部分不法厂商在保健食品中借机添加违法的化学药物[9],增加了患者服用药物剂量和服用种类的不确定性和随机性,由此带来的一系列不良反应,轻则损伤脏器,重则危及生命[10‒11]。为保证保健食品安全生产和质量控制,除了要有相关法律文件作为监管依据外,还要求具有灵敏快速的检测方法。目前保健食品中非法添加检测方法主要有红外光谱或紫外分光光度法[12‒14]、薄层色谱法[15‒16]、液相色谱法[17‒19]、色谱-质谱联用法[20‒22]、电化学方法[23]、离子迁移谱[24‒25]等。然而降三高类药物种类繁多,这些方法对保健食品基质中化学药物的检测并不全面且精确度较低,笔者通过对茶剂、软胶囊剂、硬胶囊等6种保健食品样品剂型进行处理优化,利用高分辨质谱的高分辨率、高灵敏的特性建立了以精准质量数、二级谱图为基础的保健食品中非法添加的58种降三高类药物的高效液相色谱-高分辨飞行时间质谱(HPLC-TOF/MS)检测方法,可为保健食品中降三高类非法添加物的监管提供技术支持。
1、 实验部分
1.1 主要仪器与试剂
飞行时间高分辨质谱仪:Trilpe TOF 5600+系统,美国AB SCIEX公司。
液相色谱仪:20A型,日本岛津公司。
超纯水器:ElixMilli-Q型,美国密理博公司。
超声波清洗器:kq-600型,中国昆山市超声仪器有限责任公司。
电子分析天平:AL204型,感量为0.001 mg,瑞士梅特勒-托利多公司。
涡旋混匀器:Vortex-Genie型,美国科学产业公司。
色谱柱:Kinetex F5型,美国飞诺美公司。
高速冷冻离心机:美国贝克曼公司。
有机相微孔滤膜:0.22 μm,美国沃特世公司。
甲醇、乙腈、正己烷:色谱纯,美国赛默飞世尔科技公司。
甲酸:优级纯,美国ROE scientific inc公司。
甲酸铵:分析纯,天津市光复精细化工研究所。
N-丙基乙二胺(PSA)、十八烷基二氧化硅(C18)和石墨化炭黑(GCB):分析纯,天津博纳艾杰尔科技有限公司。
无水硫酸镁:分析纯,国药集团化学试剂有限公司。
氯化钠:分析纯,天津市博迪化工股份有限公司。
柠檬酸钠:分析纯,天津市申泰化学试剂有限公司。
混合标准溶液:(1) 甲醇中6种药物混合标准溶液,利多卡因、盐酸可乐定、盐酸美西律、盐酸普罗帕酮、盐酸维拉帕米、利血平的质量浓度均为10 μg/mL;(2) 甲醇中11种降压类药物混合标准溶液,阿替洛尔、盐酸可乐定、卡托普利、哌唑嗪、利血平、硝苯地平、氨氯地平、尼群地平、尼莫地平、非洛地平、氢氯噻嗪的质量浓度均为100 μg/mL;(3) 甲醇中15种药物混合标准溶液,比索洛尔、艾司洛尔、布曲嗪、苯乙双胍、福莫特罗、酚妥拉明、瑞舒伐他汀钙、洛沙坦、沙美特罗、替宁酸、吲哚洛尔、伊拉地平、伊索唑胺、磷酸苯丙哌林、苯乙醇胺A的质量浓度均为50 μg/mL;(4) 乙腈中5种药物混合标准溶液,坎利酮、盐酸马布特罗、培哚普利、瑞格列奈、西地那非的质量浓度均为50 μg/mL;(5) 乙腈中洛非西定标准溶液,100 μg/mL;(6) 乙腈中地尔硫卓标准溶液,100 μg/mL;(7) 甲醇中13种药物混合标准溶液,盐酸克伦潘特、卡维地洛、磷酸利美尼定、N-乙酰-2-苄基色胺、溴布特罗、硫酸异丙喘宁、二羟丙茶碱、菲诺特罗、利多氟嗪、盐酸吡格列酮、羟甲基克伦特罗、阿西莫司、阿佐塞米的质量浓度均为50 μg/mL;(8) 乙腈中4种药物混合标准溶液,辛伐他汀、洛伐他汀、美伐他汀、去羟基洛伐他汀的质量浓度均为100 μg/mL;(9) 甲醇中21种降糖药物混合标准溶液,其中达格列净、坎格列净、曲格列酮、环格列酮的质量浓度均为50 μg/mL,西他列汀、甲苯磺丁脲、妥拉磺丁脲、维达列汀、格列波脲、那格列奈、格列喹酮、莫格他唑、盐酸吡格列酮、罗格列酮、氯磺丙脲、格列吡嗪、醋磺己脲、瑞格列奈、格列苯脲、格列美脲、卡达林的质量浓度均为1 μg/mL;(10) 甲醇中13种降糖药物混合标准溶液,甲苯磺丁脲、格列苯脲、格列齐特、格列吡嗪、格列喹酮、格列美脲、马来酸罗格列酮、瑞格列奈、盐酸吡格列酮、盐酸二甲双胍、盐酸苯乙双胍、盐酸丁二胍、格列波脲的质量浓度均为100 μg/mL;天津阿尔塔科技有限公司。
保健食品样品:委托检测样品。
实验用水为超纯水,由ElixMilli-Q型超纯水器过滤而得。
1.2 溶液配制
41种药物混合标准储备液:分别取甲醇中6种药物混合标准溶液0.5 mL,甲醇中15种药物混合标准溶液、乙腈中5种药物混合标准溶液、甲醇中13种药物混合标准溶液、甲醇中21种降糖药物混合标准溶液各0.1 mL,甲醇中11种降压类药物混合标准溶液、乙腈中洛非西定标准溶液、乙腈中地尔硫卓标准溶液、乙腈中4种药物混合标准溶液、甲醇中13种降糖药物混合标准溶液各0.05 mL,配制成41种药物质量浓度均为500 ng/mL的混合标准储备液。
17种药物混合标准储备液:取甲醇中21种降糖药物混合标准溶液适量,将其中质量浓度为1 μg/mL的17种药物配制成质量浓度均为500 ng/mL的混合标准储备液。
58种药物系列混合标准工作溶液:以0.1%甲酸溶液为稀释剂,将上述两种混合标准储备液配制成58种药物的质量浓度均分别为0.001、0.01、0.1、0.2、0.5、1、2、5、10、20、50、100、200、500 ng/mL的系列混合标准工作溶液。
0.5%酸化乙腈溶液:取500 μL甲酸,用乙腈定容至100 mL,得到体积分数为0.5%酸化乙腈溶液。
空白样品溶液:将空白样品按照样品处理程序处理,得空白样品溶液。
加标样品溶液:向空白样品中加入58种物质混合标准储备液,并依照样品处理程序处理,得到各药物添加水平均为50 ng/mL的加标样品溶液。
1.3 样品处理
1.3.1 提取
液体剂(养生酒):准确量取0.5 g样品,用酸化乙腈-水(体积比为9∶1)溶液定容至10 mL,混合均匀,以10 000 r/min转速离心3 min,得上清液待净化。
固体剂(代用茶、滴丸、硬胶囊、压片):准确称取0.5 g经研磨的固体样品,加入1 mL水浸泡10 min,加入9 mL酸化乙腈,涡旋混匀,超声提取10 min,加入1 g MgSO4、0.9 g NaCl、0.2 g Na3C6H5O7,涡旋混匀,以10 000 r/min转速离心3 min,上清液待净化。
油剂(软胶囊):准确称取0.5 g样品,加入2 mL饱和酸化乙腈正己烷,涡旋使其溶解,加入10 mL饱和正己烷酸化乙腈提取,涡旋混匀,以10 000 r/min转速离心3 min,取乙腈层待净化。
1.3.2 净化
取1 mL提取液至2 mL EP管中,加入20 mg PSA、20 mg C18、2 mg GCB,涡旋振荡2 min,以10 000 r/min转速离心3 min,取上层清液,经0.2 μm滤膜得滤液,作为样品净化液。
1.4 仪器工作条件
1.4.1 色谱仪
色谱柱:Kinetex F5柱(3.0 mm×100 mm,2.6 μm,美国飞诺美公司);流动相:A相为0.1%甲酸-5 mmol/L甲酸铵水溶液,B相为甲醇-乙腈(体积比为1∶1)溶液,流量为0.25 mL/min;洗脱方式:梯度洗脱,洗脱程序见表1;柱温:30 ℃;进样体积:10 μL。
表1 梯度洗脱程序
Tab. 1 Program of gradient elution
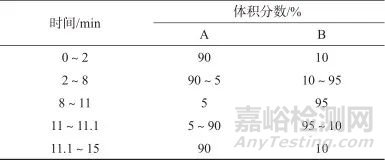
注:A—0.1%甲酸-5 mmol/L甲酸铵水溶液,B—甲醇-乙腈(体积比为1∶1)溶液。
1.4.2 质谱仪
离子源:电喷雾电离(ESI)源;扫描方式:正离子扫描;离子源温度:550 ℃;喷雾电压:5 500 V;一级扫描模式:质谱扫描范围(TOF)为m/z 100~1 200,去簇电压为60 V,碰撞能量为10 eV;二级扫描模式(IDA-MS/MS):扫描范围(IDA)为m/z 50~1 200,去簇电压为60 V,碰撞能量为20~60 eV;气帘气(CUR)压力:241 kPa;雾化气(GAS1)压力:379 kPa;辅助加热气压力(GAS2):379 kPa。根据各类物质结构特征,选择正离子模式,除达格列净和坎格列净在正离子模式下形成[M+NH4]+准分子离子峰,其余物质均形成[M+H]+准分子离子峰。58种违禁降三高药物的质谱参数见表2。
表2 58种降三高药物信息及质谱参数
Tab. 2 Information and mass spectrometry parameters of 58 drugs for reducing three highs
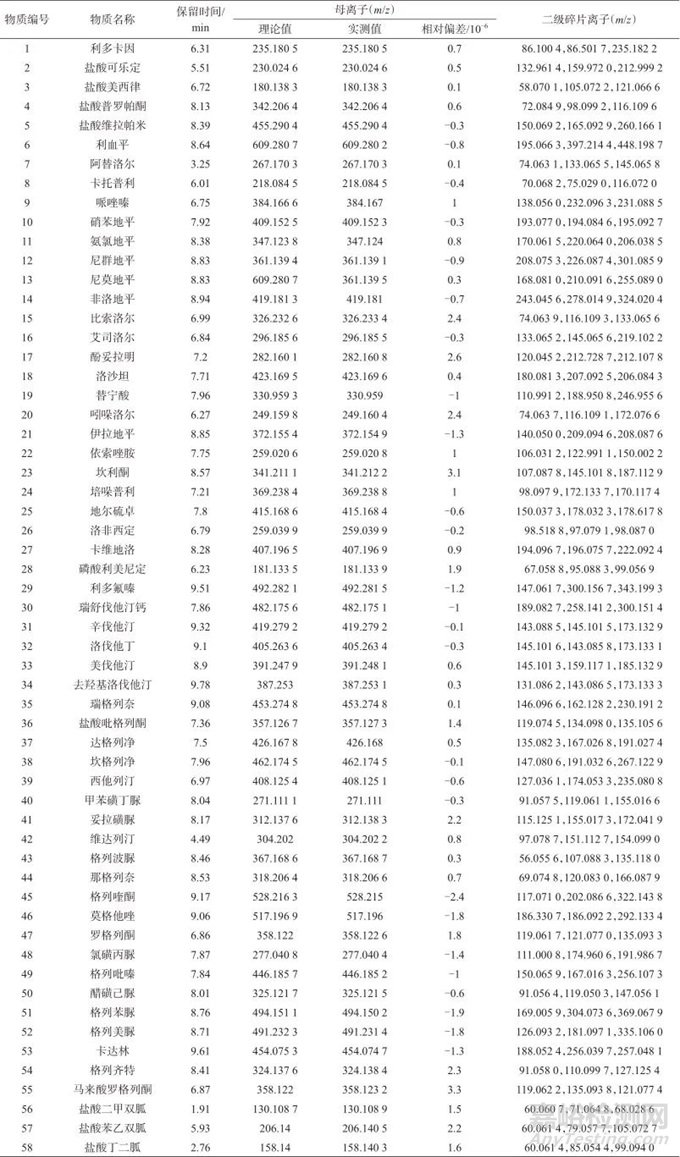
注:编号1~29为降压类药物;编号30~34为降脂类药物;编号35~58为降糖类药物。
1.5 定性定量数据库建立
利用HPLC-TOF/MS技术与SCIEX-OS-Q软件,建立包括58种常见降三高类物质的一级、二级谱图数据库,建立快速定性、定量筛查方法。在实际样品筛查时,不使用标准品对照,直接利用一级精准质量数据库和二级碎片离子谱库进行初步定性和定量筛查,实现快速高通量筛查。
1.6 样品测定
准确称取各类剂型样品0.5 g,按照1.3方法进行样品处理,溶液稀释至1 000倍体积,过0.2 μm滤膜得滤液,在1.4仪器工作条件下测定,利用一级、二级谱图数据库进行定性和定量。
2、 结果与讨论
2.1 提取条件优化
参考国家标准SN/T 5357—2021《出口保健食品中多类非法添加物的测定 液相色谱—质谱/质谱法》对样品处理过程进行优化,由于提取剂等试剂对不同剂型样品的影响差别不大,所以主要以茶剂(固体剂)为例进行样品处理方法的优化。
2.1.1 提取溶剂选择
用甲酸对甲醇和乙腈进行酸化,甲酸体积分数均为0.5%,对比酸化甲醇和酸化乙腈作为提取剂时各目标物回收率情况。每种提取剂设置3次平行试验,标准偏差为0.32%~14.55%,两种提取剂的提取效率如图1所示。由图1可以看出,以酸化乙腈作为提取剂时各物质回收率较好。

图1 不同提取溶剂时茶剂中58种药物的提取效率
Fig. 1 Extraction efficiency of 58 drugs in tea preparations at different extraction solvents
从整体来看,乙腈的提取效率优于甲醇,所以,在对液体剂和油剂类样品进行提取时,也使用乙腈进行提取。液体剂类样品使用0.5%酸化乙腈-水(体积比为9∶1)进行提取;油剂类样品用正己烷与0.5%酸化乙腈互为饱和的溶液(正己烷与0.5%酸化乙腈体积比为1∶5),将上层饱和酸化乙腈正己烷作为除油剂,将下层饱和正己烷酸化乙腈作为的提取剂。
2.1.2 盐析剂用量选择
固体样品提取时选用无水MgSO4、NaCl和Na3C6H5O7作为盐析剂,无水MgSO4可除去有机相中的水,并且可萃取出极性杂质干扰物,如糖类等;NaCl和Na3C6H5O7可控制提取剂极性,但不易过量[26‒27]。对加标茶剂样品进行试验考察,当MgSO4、NaCl和Na3C6H5O7加入量分别为2、0.5、0.5 g和1、0.2、0.2 g时对比各目标物的回收率。不同盐析剂加入量设置3次平行试验,标准偏差为0.51%~14.69%,盐析剂不同用量时的提取效率如图2所示。由图2可以看出,当盐析剂MgSO4、NaCl、Na3C6H5O7加入质量分别为2、0.5、0.5 g时,有13种目标物回收率明显降低,分别是编号为1、2、3、4、6、7、11、20、21、25、33、40、57的目标物,尤其是编号为7、57的目标物,其回收率低于60%,因此确定盐析剂MgSO4、NaCl、Na3C6H5O7的用量分别为1、0.2、0.2 g。
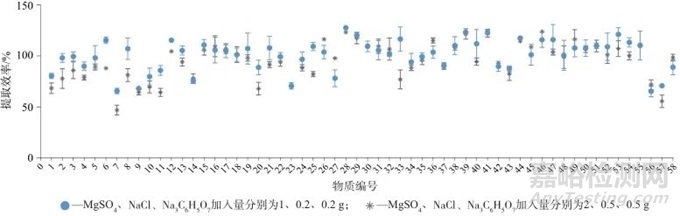
图2 盐析剂不同用量时茶剂中58种药物的提取效率
Fig. 2 Extraction efficiency of 58 drugs in tea preparations at different dosage of salting-out agent
2.1.3 除油剂用量选择
软胶囊样品基质主要成分是油脂,选择饱和酸化乙腈正己烷作为除油剂,正己烷饱和酸化乙腈作为提取剂。对除油剂用量进行优化,对比除油剂用量分别为1 mL和2 mL时各药物回收率,两种加入量设置3次平行试验,标准偏差为0.41%~14.64%,除油剂不同用量时的提取效率如图3所示。由图3可以看出,当除油剂用量为1 mL时,有20种目标物的回收率明显降低,分别是编号为2、4、5、6、7、8、9、11、13、18、19、22、26、44、48、50、55、56、57和58的目标物,尤其是编号为8和50的目标物,其回收率低于60%,因此确定除油剂用量为2 mL。
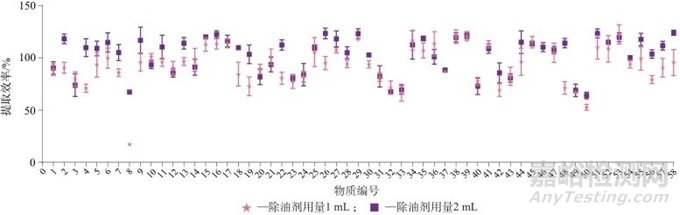
图3 除油剂不同用量时油剂中58种药物的提取效率
Fig. 3 Extraction efficiency of 58 drugs in oil agents at different dosages of degreasers
2.2 净化技术优化
由于部分保健食品基质容易对待测物产生干扰,因此需要对样品提取液进行净化。采用基于QuEChERS原则的样品处理技术,选择PSA、C18、GCB为吸附剂,PSA对极性物质具有吸附作用,可去除糖类、有机酸等干扰;C18对非极性物质具有吸附作用,可去除脂质、固醇等干扰;GCB可有效去除色素等干扰,但影响平面结构物质的回收率[28‒29]。分别考察3种吸附剂在不同用量时各药物的回收率,3种吸附剂的用量分别为PSA:20、50、100 mg;C18:20、50、100 mg;GCB:0、2、5、10 mg,图4~图6分别为3种吸附剂不同用量时58种药物的平均回收率。结果显示,受PSA影响较大的有编号为8、19、20、24、52、56、58的目标物;受C18影响较大的有编号为31、34、57、58的目标物;受GCB影响较大的有编号为1、4、5、6、8、9、10、11、14、20、22、23、27、29、35、36、45、49、51、52、53的目标物。综合考虑,确定吸附剂PSA、C18、GCB的用量分别为20、20、2 mg。
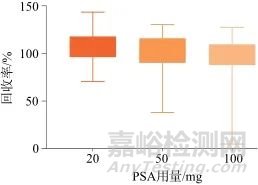
图4 PSA不同用量时58种目标物的平均回收率
Fig. 4 Average recoveries of 58 target substances at different dosage of PSA
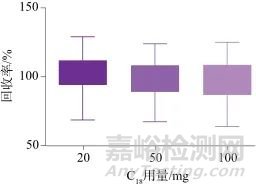
图5 C18不同用量时58种目标物的平均回收率
Fig. 5 Average recoveries of 58 target substances at different dosage of C18
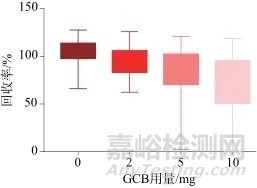
图6 GCB不同用量时58种目标物的平均回收率
Fig. 6 Average recoveries of 58 target substances at different dosage of GCB
2.3 流动相选择
以混合标准溶液为分析对象,分别选择4种流动相体系进行试验,(1) A相为0.1%甲酸-5 mmol/L甲酸铵溶液,B相为乙腈;(2) A相为0.1%甲酸-5 mmol/L甲酸铵溶液,B相为乙腈-0.1%甲酸-5 mmol/L甲酸铵溶液;(3) A相为0.1%甲酸-5 mmol/L甲酸铵溶液,B相为甲醇+乙腈(体积比为1∶1)溶液;(4) A相为0.1%甲酸溶液,B相为乙腈。结果表明,盐酸可乐定在(1)、(3)流动相体系中具有较好的峰形,在(2)、(4)流动相体系中峰形变宽且前延峰明显;阿替洛尔在4种流动相中均出现杂峰,在(2)、(4)流动相体系中于2 min之前出峰,在(4)流动相体系中出现双峰且均能匹配;甲苯磺丁脲在4种流动相中均出现杂峰,但在(4)流动相体系中有所改善且灵敏度提高;维达列汀在4种流动相中均出现杂峰,在(2)、(4)流动相体系中峰形变差且出现拖尾现象;罗格列酮在4种流动相中均出现双峰且均能匹配;盐酸二甲双胍在4种流动相中均出现拖尾现象,但(4)流动相体系中峰形有所改善但灵敏度降低。总体来看绝大多数物质在(3)流动相体系中具有最高的灵敏度,且具有较好的峰形,故选择(3)流动相体系,即A相为0.1%甲酸溶液-5 mmol/L甲酸铵溶液,B相为甲醇-乙腈(体积比为1∶1)溶液。
2.4 色谱图
混合标准溶液、空白样品溶液加标样品溶液色谱图如图7~图9所示。
图7 混合标准溶液提取离子色谱图
Fig. 7 Extraction ion chromatogram of mixed standard solution
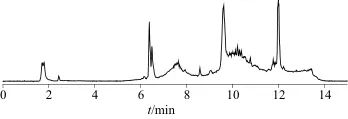
图8 空白样品溶液总离子流色谱图
Fig. 8 Total ion current chromatogram of blank sample solution
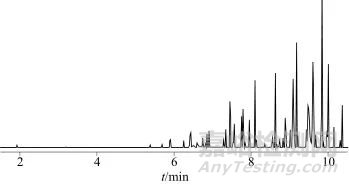
图9 加标样品溶液提取离子色谱图
Fig. 9 Extraction of ion chromatogram of spiked sample solution
2.5 线性方程、检出限和定量限
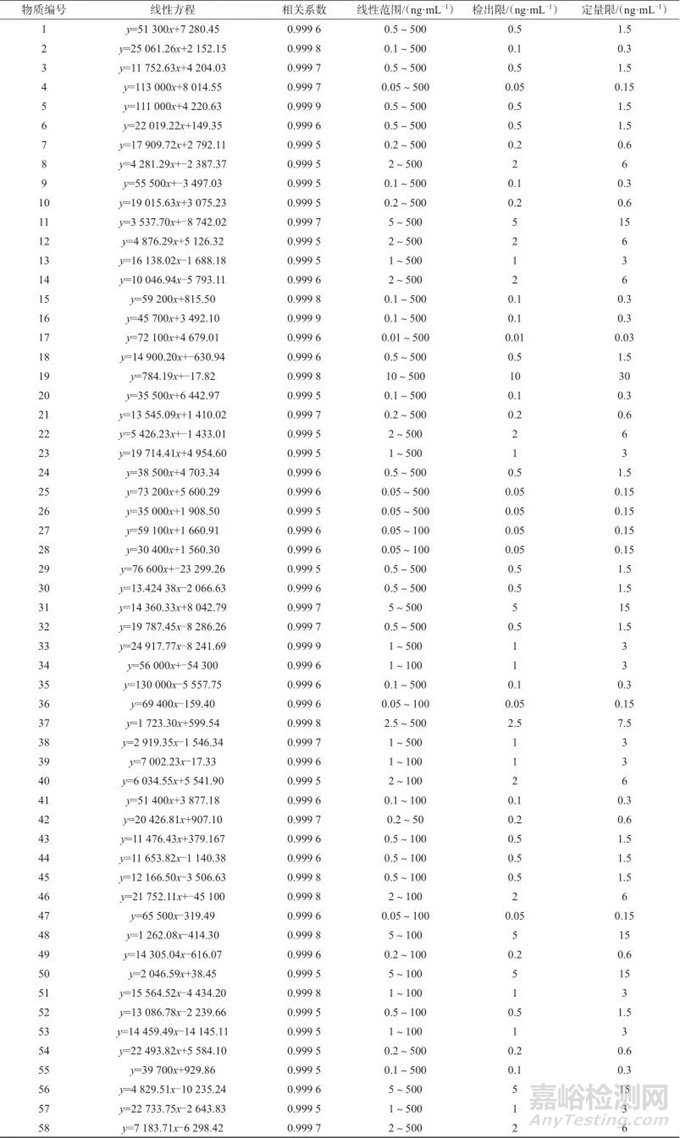
在1.4仪器工作条件下,分别测定58种药物系列混合标准工作溶液,以标准溶液质量浓度x为横坐标,以色谱峰面积y为纵坐标,得到各目标物的回归方程。逐级稀释58种药物混合标准溶液,分别以3倍信噪比和10倍信噪比对应的质量浓度作为方法检出限和定量限。58种药物质量浓度线性范围、线性方程、相关系数、检出限和定量限见表3。由表3可知,各目标物线性关系良好,相关系数均大于0.999 0,58种药物的检出限为0.01~10 ng/mL,定量限为0.03~30 ng/mL。表明该方法具有较高的灵敏度。
表3 质量浓度线性范围、线性方程、相关系数、方法检出限和定量限
Tab. 3 Linear range of mass concentration、linear equation、correlation coefficient、method detection limit and quantitative limit
2.6 加标回收与精密度试验
向空白样品中加入58种药物混合标准储备液,使卡托普利、替宁酸、盐酸丁二胍、氯磺丙脲、醋磺己脲的加标质量浓度均为25 ng/mL,其余药物的加标质量浓度均为5 ng/mL,按照1.3样品处理方法,平行处理6份样品溶液,在1.4仪器工作条件下分别进行测定,结果见表4。由表4可知,58种目标物加标平均回收率为64.25%~115.3%;测定结果的相对标准偏差为1.1%~17.85%,表明该方法准确度、精密度均良好。
表4 加标回收与精密度试验结果(n=6)
Tab. 4 Standard addition recoveries and precision test results (n=6) ( % )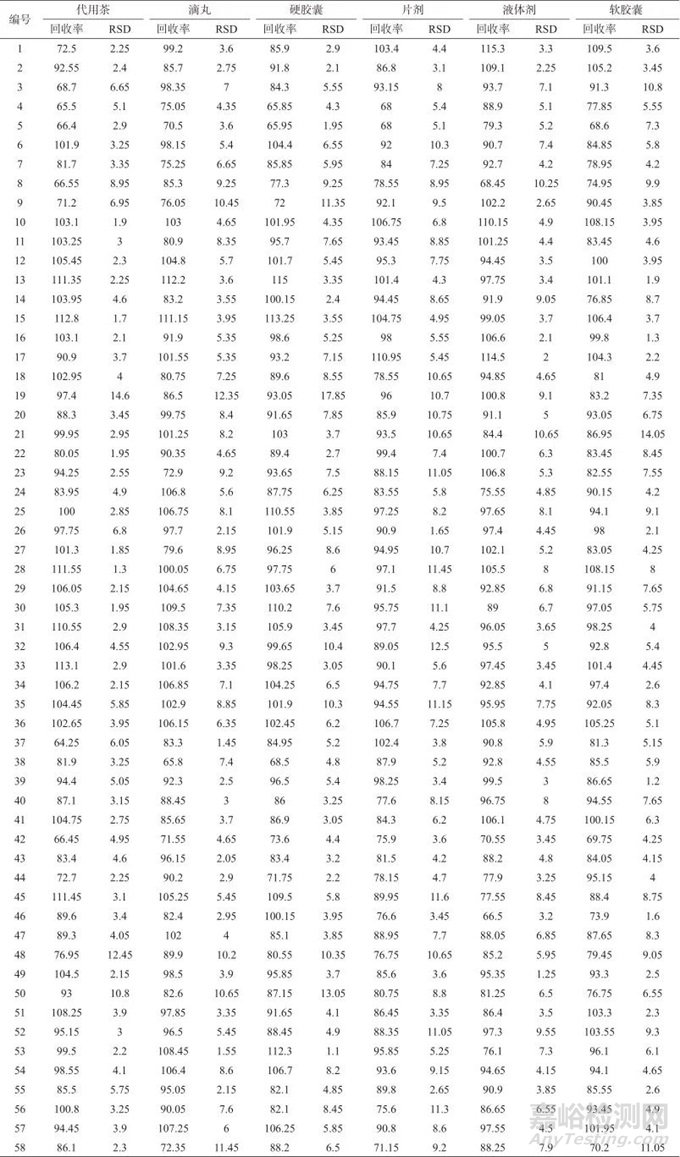
注:卡托普利、替宁酸、盐酸丁二胍、氯磺丙脲、醋磺己脲的加标质量浓度均为25 ng/mL,其余药物的加标质量浓度均为5 ng/mL。
2.7 稳定性试验
按1.3方法制备样品溶液,分别在第0、2、4、8、12、24 h进样测定,各目标物色谱峰面积测定结果的相对标准偏差为0.91%~8.95%,说明各药物样品溶液在24 h内稳定性良好。
3、 结语
利用HPLC-TOF/MS联用技术以及SCIEX-OS-Q分析软件建立了针对降三高类保健食品中违禁添加成分的检测方法,对6种剂型的常见保健食品样品进行优化处理,对样品提取液进行净化,在保证回收率的同时,有效降低杂质干扰。通过精准分子质量数据库和碎片离子谱库的建立,实现快速、高精确度、更全面的物质筛查,可在15 min内同时对58种物质进行定性、定量分析,有助于对保健食品违禁添加物的监管与治理。为探究保健食品中更多降三高类药物的非法添加,还需要不断完善数据库,以提供更好的技术支撑。
参考文献
1 马东红,王志信,吕玉敏,等.糖尿病肾脏疾病与其他慢性肾脏疾病腹膜透析患者临床特点及心血管疾病患病率和危险因素的对比分析[J].中国糖尿病杂志,2020,28(12):903.
MA Donghong,WANG Zhixin,LYU Yumin,et al. Clinical characteristics,prevalence and risk factors for cardiovascular diseases in peritoneal dialysis[J]. Chinese Journal of Diabetes,2020,28(12):903.
2 BLOOMFIELD G S,YINKO S. A series of unhealthy developments the multiple facets of cardiovascular risk factors in China[J]. Journal of the American College of Cardiology,2016,68(8):834.
3 徐春晓,郭晓雷,鹿子龙,等. 2016~2020年山东省35~75岁缺血性心血管病患者“三高”控制及二级预防用药状况分析[J].中国慢性病预防与控制,2023,31(5):343
XU Chunxiao,GUO Xiaolei,LU Zilong,et al. Control of “three highs” and use of secondary prevention drugs in patients (35-75 years old) with ischemic cardiovascular diseases in Shandong Province from 2016 to 2020[J]. Chinese Journal of Prevention and Control of Chronic Diseases,2023,31(5):343.
4 蔡晓琪,高欣蕾,王庭俊,等.异常代谢组分积聚与体位性低血压的关系[J].中华高血压杂志,2020,28(11):1 051.
CAI Xiaoqi,GAO Xinlei,WANG Tingjun,et al. Relationship between the accumulation of metabolic syndrome components and orthostatic hypotension[J]. Chinese Journal of Hypertension,2020,28(11):1 051.
5 LIMA N K C,ABBASI F,LAMENDOLA C,et al. Prevalence of insulin resistance and related risk factors for cardiovascular disease in patients with essential hypertension[J]. American Journal of Hypertens,2009,22(1):106.
6 LASTRA G,SYED S,KURUKULASURIYA L R,et al. Type 2 diabetes mellitus and hypertension:an update[J]. Journal of Clinical Endocrinology & Metabolism,2014,43(1):103.
7 林永阳.高血压患者联合用药的合理性与不良反应特点及联合用药禁忌证[J].临床合理用药杂志,2018,11(18):105.
LIN Yongyang. The rationality of combined medication in patients with hypertension,the characteristics of adverse reactions and the contraindications of combined medication[J]. Chinese Journal of Clinical Rational Drug Use,2018,11(18):105.
8 ANTONOPOULOS N,MACHAIRAS G,MIGIAS G,et al. Hydrophilic interaction liquid chromatography-electrospray ionization mass spectrometry for therapeutic drug monitoring of metformin and rosuvastatin in human plasma[J]. Molecules,2018,23(7):1 548.
9 贾婧怡,李玮,张烨,等.保健食品中非法添加药物事件及检测技术[J].食品安全质量检测学报,2020,11(11):3 558.
JIA Jingyi,LI Wei,ZHANG Ye,et al. Illegal drug addition events and determination technologies in health supplements[J]. Journal of Food Safety & Quality,2020,11(11):3 558.
10 DASGUPTA K,QUINN R R,ZARNKE K B,et al. The 2014 Canadian hypertension education program recommendations for blood pressure measurement,diagnosis,assessment of risk,prevention,and treatment of hypertension[J]. Canadian Journal of Cardiology,2014,30(5):485.
11 SUN Mingyang,YUAN Tao,WAN Mingchen,et al. Optimal statin use for prevention of sepsis in type 2 diabetes mellitus[J]. Diabetology & Metabolic Syndrome,2023,15(1):75.
12 许凤,冯钰,付双,等.薄层色谱-显微红外光谱(TLC-MicroIR)联用技术检测减肥保健食品中非法添加盐酸芬氟拉明[J].中国医院药学杂志,2016,36(5):358.
XU Feng,FENG Yu,FU Shuang,et al. Determination of fenfluramine hydrochloride added illegally into anti-obesity and healthcare food by TLC-MicroIR method[J] Chinese Journal of Hospital Pharmacy,2016,36(5):358.
13 刘佳明,陈立波,吕维维,等.近红外光谱快速鉴别清咽类制剂中3种非法添加化学药物的方法[J].食品与药品,2023,25(1):51.
LIU Jiaming,CHEN Libo,LYU Weiwei,et al. Rapid identification of 3 illegal added chemical drugs in qingyan preparations by near infrared spectrum[J]. Food and Drug,2023,25(1):51.
14 宋翔,王云贵,黄登宇.硝苯地平免疫胶体金探针的制备[J].食品安全质量检测学报,2016,7(8):3 350.
SONG Xiang,WANG Yungui,HUANG Dengyu. Preparation of nifedipine immune colloidal gold probe[J]. Journal of Food Safety & Quality,2016,7(8):3 350.
15 ROJANARATA T,PLIANWONG S,SU-UTA K,et al. Electrospun cellulose acetate nanofibers as thin layer chromatographic media for eco-friendly screening of steroids adulterated in traditional medicine and nutraceutical products[J]. Talanta,2013,115:208.
16 PATEL K G,SHAH P M,SHAH P A,et al. Validated high performance thin-layer chromatographic (HPTLC) method for simultaneous determination of nadifloxacin,mometasone furoate,and miconazole nitrate cream using fractional factorial design[J]. Jornal of Food and Drug Analysis,2016,24(3):610.
17 KOTHA R R,LUTHRIA D L. Curcumin:biological,pharmaceutical,nutraceutical,and analytical aspects[J]. Molecules,2019,24(16):2 930.
18 HANIF S,SYED M A,RASHID A J,et al. Validation of a novel RP-HPLC technique for simultaneous estimation of lignocaine hydrochloride and tibezonium iodide:greenness estimation using AGREE penalties[J]. Molecules,2023,28(8):3 418.
19 MEHMOOD T,HANIF S,AZHAR F,et al. HPLC method validation for the estimation of lignocaine HCl,ketoprofen and hydrocortisone:greenness analysis using AGREE score[J]. International Journal of Molecular Sciences,2023,24(1):440.
20 ALVES R D,ROMERO-GONZALEZ R,LOPEZ-RUIZ R,et al. Fast determination of four polar contaminants in soy nutraceutical products by liquid chromatography coupled to tandem mass spectrometry[J]. Analytical and Bioanalytical Chemistry,2016,408(28):8 089.
21 JIN Yin,HE Chaoqun,DI Xiangjie,et al. Simultaneous determination of lidocaine and its active metabolites in plasma by UPLC-MS/MS and application to a clinical pharmacokinetic study in liver cancer patients with laparoscopic hepatectomy[J]. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences,2022,1 207:123 362.
22 STEPANOVA E S,BARSEGYAN S S,MAKARENKOVA L M,et al. Development and validation of a quantitative determination method for the antitubercular drug PBTZ169 in biological media[J]. Journal of Tropical Pharmacy and Chemistry,2018,52(3):248.
23 SIRIVIBULKOVIT K,WILAIRAT P,NACAPRICHA D,et al. A simple cost-effective paper-based electrochemical device for detection of adulterated sibutramine in slimming products[J]. Analytical Methods,2022,14(25):2 461.
24 冯雪,尹利辉,金少鸿,等.离子迁移谱法快速检测保健食品中添加的5型磷酸二酯酶抑制剂[J].药物分析杂志,2016,36(2):313.
FENG Xue,YIN Lihui,JIN Shaohong,et al. Rapid detection of undeclared phosphodiesterase type 5 inhibitors indietary supplements by ion mobility spectrometry[J]. Chinese Journal of Pharmaceutical Analysis,2016,36(2):313.
25 LI Lingfeng,WANG Tiesong,HAN Ke,et al. Rapid identification of illegally addition in traditional Chinese antidiabetic medicine by field asymmetric ion mobility spectrometric technique[J]. Chinese Journal of Analytical Chemistry,2014,42(4):519.
26 PERESTRELO R,SILVA P,PORTO-FIGUEIRA P,et al. QuEChERS-Fundamentals,relevant improvements,applications and future trends[J]. Analytica Chimica Acta,2019,1 070:1.
27 REJCZAK T,TUZIMSKI T. A review of recent developments and trends in the QuEChERS sample preparation approach[J]. Open Medicinal Chemistry Journal,2015,13(1):980.
28 HUANG Yousheng,SHI Ting,LUO Xiang,et al. Determination of multi-pesticide residues in green tea with a modified QuEChERS protocol coupled to HPLC-MS/MS[J]. Food Chemistry,2019,275:255.
29 GONZALEZ-CURBELO M A,SOCAS-RODRIGUEZ B,HERRERA-HERRERA A V,et al. Evolution and applications of the QuEChERS method[J]. Trac-Trends in Analytical Chemistry,2015,71:169.
Determination of 58 illegally reducing blood pressure, blood lipids and blood glucose substances in health care products by high performance liquid chromatography-high resolution mass spectrometry

来源:化学分析计量


