您当前的位置:检测资讯 > 行业研究
嘉峪检测网 2024-07-18 09:16
摘要
罕见病是全人类面临的重大医学挑战。目前全球范围内已知的罕见病有7000~10000 种,中国罕见病患者数量众多,大多数患者面临着无药可治或者境外有药、境内无药的问题。药品审评审批制度改革以来,我国从政策文件、法律规范、技术标准等维度规范了罕见病范围、罕见病药品加快审评途径、技术标准等内容,加快罕见病药品上市,以求不断满足罕见病患者用药需求。通过对我国罕见病药品审评审批制度的分析,建议从罕见病药品认定标准、设立专门机构、建立市场独占期制度、加大研发指导等方面完善罕见病药品审评审批制度。
【关键词】 罕见病;罕见病药品;审评审批制度改革
罕见病是全人类面临的重大医学挑战。罕见病患病率、发病率在总人口中占比极低,但严重威胁着患者的生命和健康。我国罕见病患者数量众多,大多数患者面临着无药可治或境外有药、境内无药的问题。世界范围内,罕见病药品也存在研发成本高昂、市场空间狭小、投资回报率低且风险高的困境,企业研发动力不足。党的二十大报告指出,必须坚持在发展中保障和改善民生,推进健康中国建设,把保障人民健康放在优先发展的战略位置。在我国罕见病总体人群数量庞大、疾病负担重,该类患者人群面临的困境正越来越引起政府和社会的广泛关注。药品审评审批制度改革以来,我国逐步构建起以临床价值为导向的科学审评体系,通过多种药品加快上市注册程序,加快罕见病药品上市,不断满足罕见病患者用药需求。
一、罕见病及罕见病药品
罕见病是一种患病率和发病率极低的疾病,由于经济发展不同和种族差异等因素,各国对罕见病定义有微小差别,但是有相对共同、经典的核心病种。世界目前全球范围内已知的罕见病有7000~10000种,其中公认的约7000 多种,占人类已知疾病的10% ,涉及患者数约 3.5 亿例[1]。据2020 年一项流行病学调查研究显示,14岁及以下死亡的儿童中约56.8%患有罕见病,是儿童死亡的主要原因。约80%的罕见病为遗传性疾病,多数在生命早期即可发病。有数据显示,约35%的罕见病患儿在出生后1 年内死亡。
1.1 境外罕见病定义
由于世界各国和地区经济发展、医疗水平、社会保障程度差异较大,目前对于罕见病未有统一的标准。世界卫生组织( Wond Health Organization,WHO)将罕见病定义为发病率在 0.65%~1%的疾病。美国是世界上最早提出罕见病并从法律层面进行界定的国家,其主要基于患者数对罕见病进行定义,在1983 年出台的《孤儿药法案》(0rphan Drug Act,ODA)中将“美国患病人数少于 20 万的疾病”定义为罕见病,2003 年发布的《罕见病法案》( Rare Dis.eases Act,RDA)对罕见病定义进行了扩展:“患病人数少于 20 万的疾病,或者患病人数超过 20 万但预期其治疗药品销售额难以收回研发成本的疾病”。据此,美国 FDA 在 2013 年发布的“0rphanDrug Regulations”中,明确孤儿药是指在美国影响人Drug Regulations”中,明确孤儿药是指在美国影响人数少于 20 万例,或影响人数多于 20 万例且没有合理预期表明在美国的销售收人大于研发成本的所有疾病的药品。澳大利亚也采用患病人数标准定义罕见病(患者少于2000 例的疾病)。欧盟主要以患病率为标准进行定义,规定患病率低于 0.5%、危及生命或严重慢性衰退性疾病,其针对性治疗药品的销售额难以收回研发成本的疾病,或目前该疾病没有令人满意的治疗药品的疾病为罕见病。俄罗斯、新西兰、巴西等国家也采用此定义标准。还有一些国家和地区采用患病人数和患病率相结合的标准,如日本规定,患病人数少于5 万或患病率低于 1/2500的疾病,且目前无合适的用于预防和诊疗药品的疾病为罕见病。韩国亦采用此种定义标准。不难看出,各个国家和地区在定义罕见病中主要包含患病率、患者总人口、疾病危重症3个因素,有的采用1个标准,有的采用多个标准。但在定义罕见病用药时,均将经济因素,即药品销售收人与研发投人成本之间的关系纳人考量范围。
1.2 我国罕见病定义
自 1984年“罕见病”概念在我国出现至今,目前在法律法规等规范性文件中,尚未对罕见病进行明确界定。2010年,中华医学会医学遗传学分会提出将罕见病定义为“患病率<1/500 000 或新生儿发病率<1/10 000 的疾病”。《中国罕见病定义研究报告 2021》指出,新生儿发病率<1/10 000、患病率<1/10 000、患者数<14 万的疾病被定义为罕见病。目前对罕见病界定的文件是2018年5月11日国家卫生健康委员会等五部委联合发布的《第一批罕见病目录》[2],通过目录的方式列举了 121 种罕见病。国家卫生健康委员会等机构此前公布的数据显示我国现有各类罕见病患者2000多万例,每年新增患者超过 20 万例,95%的患者缺少特效药治疗[3]。2023 年9月18 日国家卫生健康委员会等6个部门联合制定了《第二批罕见病目录》列举了89 种罕见病。目前2批目录通过列举的方式,划定了207种罕见病范围。罕见病定义的不断清晰,有利于我国罕见病的诊疗和保障走向规范化、制度化和法制化。2018年,国家卫生健康委员会发布《罕见病目录制订工作程序》,指出将分批遴选目录覆盖病种对目录进行动态更新;更新时间原则上不短于2年。纳人目录的病种应同时满足以下条件:① 国内外有证据表明发病率或患病率较低。②对患者和家庭危害较大。③ 有明确诊断方法。④ 有治疗或干预手段、经济可负担,或尚无有效治疗或干预手段但已纳人国家科研专项。
1.3 罕见病药品
用于预防、诊断、治疗罕见病的药物统称“罕见病药品”。世界上最早从法律层面界定罕见病的是美国,1983 年出台《孤儿药法案》,同时罕见病药品研发成本高昂、市场需求小、投资回报低,企业生产研发意愿不高,罕见病患者可用药品极其缺少,因此罕见病药品又被称为“孤儿药”(orphan drug)。在我国““孤儿药”最早出现在 1990年孙国凤等的《大阪大学克隆分泌胃酸的质子泵以探索抗溃疡剂的作用》中,在翻译时将“orphan drug”翻译为“孤儿药”。此后“孤儿药”在我国学界得到使用。目前我国的法律法规和规范性文件中一般使用“罕见病药品”或“罕见病用药”的表述[4]。
二、美国、欧盟罕见病药品审批情况
2.1 美国 FDA
美国是世界上首个针对罕见病药品进行立法的国家。1983 年,美国颁布了《孤儿药法案》,从立法层面对罕见病药品的鼓励研发、加快审评及税费减免等激励政策予以规定。此后该法案进行了4次重大修改,分别于 1985 年修订了孤儿药市场独占期条款,1997 年增加孤儿药审评费用条款,2007 年增加豁免孤儿药处方药或生物制品批准或许可的产品费及场地费条款,2017 年修订临床试验税收抵免条款[5]。美国 FDA 成立了专门机构——孤儿药研发办公室(Office of 0rphan Products Development,00DP)负责制定与修订孤儿药政策、实施孤儿药各项条例并负责孤儿药资格的认定。
美国未对孤儿药设立专门的加快审批程序。但因孤儿药的特殊身份和其临床价值,可适用美国FDA 优先审评、加速审评、快速通道、突破性治疗等加快程序。通过加快上市程序,最快的孤儿药审评审批用时缩短为6个月。孤儿药法案实施以来,罕见病药品研发上市数量显著增加。截至 2020年3月底美国 FDA 共授权 5 320 件罕见病药品资格认定,涉及2 949 种罕见病,已有 458 个罕见病药品获批上市[6]。
2.2 欧洲EMA
欧盟(European Commission,EC)于 1999 年颁布《孤儿药管理法规》为欧盟各国孤儿药法规的制定奠定了法律基础。欧洲EMA 负责统一管理欧盟范围内医疗产品有关事宜,并成立专门的孤儿药品委员会(Committee for Orphan Medical Products,COMP)负责罕见病药品孤儿药身份的认定。
获得孤儿药身份的药品由欧盟人用药品委员会(Committee for Medical Products for Human Use.CHMP)提出审评意见,可适用加速审批、有条件审批、特殊审批及优先审批等加速审批路径,并最终由EC 作出审批决定。通过审评的孤儿药可在成员国内流通与销售,无需再向成员国提交单独的上市申请。
欧盟孤儿药身份管理制度类似于美国,一方面成立了专门机构负责孤儿药的资格认定,另一方面资格认定是使用制度的前提,只有通过认定后,才可享有一系列针对孤儿药研发的激励政策,如免除监管费用、提供研发资助、提供协议援助、10 年市场独占期等。同时,由于罕见病自身特点和孤儿药研发的难度,欧盟内部成员国之间以及欧盟与其他国家监管机构开展临床试验合作和数据共享。
三、我国罕见病药品审评审批情况
3.1 制度体系
2015 年审评审批改革启动之后,我国药品监管部门持续深化改革,落实改革要求,逐步构建以临床价值为导向的审评审批体系,形成了政策、法律、规章、技术标准等多层次的制度规范,从罕见病认定标准、罕见病研发技术指导、罕见病药品加快上市程序、罕见病药品医疗保障、罕见病药物研发生产税收减免等多个维度予以规定,为企业投入罕见病药品的研发生产提供了动力,为罕见病药品加快上市提供了路径,为罕见病患者有药可用提供了保障,从规范层级来看,形成了国务院、中共中央办公厅、国务院办公厅发布的政策文件,全国人大常委会制定的法律,国家药品监督管理局等制定的部门规章,国家药品监督管理局药品审评中心(以下简称“药审中心”)等技术部门制定的技术标准等多层次、多维度的规范性文件体系。从规范内容来看,从鼓励研发、加快审评、医保保障、税收减免等方面,对于罕见病的范围、罕见病药品研发的技术标准、罕见病药品的认定、加快审评程序、上市后的市场独占期、增值税技术税率等方面对罕见病药品的研发、审评、使用、销售等环节予以细化明确。针对绝大多数罕见病为遗传疾病、多在儿童生长发育关键期发病的特点,药审中心完善审评标准体系,加强沟通与交流,发布多项罕见病用药、儿童用药等技术指导原则。同时,为避免重复开展临床试验、降低研发成本、促进境内外企业合作,药审中心还发布了多项境外已上市、境内未上市药品药学、临床试验等相关指导原则。
不难看出,经过近年来的努力,罕见病药品的相关制度建立逐渐完善,对于企业的鼓励力度也不断加大。
3.2 审评审批流程
依据 2020年1月颁布实施的《药品注册管理办法》,我国设立突破性治疗药物程序、附条件批准程序、优先审评审批程序、特别审批程序 4 条药品加快上市注册程序。与美国 FDA、欧洲 EMA 等相同,我国虽未对罕见病药品单独设立快速审评通道,但可将其纳入优先审评等加快程序[7],近年来罕见病药品上市速度显著加快[8-9]。以下从优先审评审批程序梳理罕见病药品的审批流程。
3.2.1 纳入优先审评的罕见病药品范围
临床亟须的短缺药品、防治重大传染病和罕见病等疾病的创新药和改良型新药,或列人国家药品监督管理局《临床急需境外新药名单》临床亟须的境外已上市境内未上市的罕见病药品,需具有明显临床价值。
3.2.2 审评审批流程
① 申请人在提出药品上市许可申请前,应当与药审中心进行沟通交流,探讨现有研究数据是否满足药品上市许可审查要求以及是否符合优先审评审批程序纳入条件等。②对于初步评估认为符合优先审评审批纳人条件的,经沟通交流确认后,申请人在提出药品上市许可申请的同时,一并提出优先审评审批申请,并提交相关支持性资料。③ 药审中心应当在接到申请后5d内对提交的优先审评审批申请进行审核,并将审核结果反馈给申请人。④ 药审中心对拟纳入优先审评审批程序的品种具体信息和理由予以公示,包括药物名称,申请人、拟定适应证(或功能主治)、申请日期、拟纳人理由等。公示5d内无异议的,正式纳人优先审评审批程序,并通知各相关方。公示期内提出异议的,异议方在5d内向药审中心提交书面意见并说明理由:药审中心在10d内另行组织论证后作出决定并通知各相关方。⑤ 药审中心对纳人优先审评审批程序的药品上市许可申请,按注册申请受理时间顺序优先配置资源进行审评。对纳人优先审评审批程序的药品上市许可申请,审评时限为 130 d,其中临床急需的境外已上市、境内未上市的罕见病药品审评时限为 70 d。⑥ 对纳人优先审评审批程序的药品上市许可申请,需要进行核查、检验和核准通用名称的,药品核查中心、药品检验机构和国家药典委员会应优先进行核查、检验和核准通用名称。⑦药审中心在收到核查结果、检验结果等相关材料后在审评时限内完成综合审评。⑧行政审批决定应当在 10 d内作出。
审评过程中发现需要与申请人进行沟通交流的,药审中心可根据具体情况优先安排。
罕见病药品也可适用突破性治疗药物程序、附条件批准程序加快审批。如果在药物临床试验期间,用于防治严重危及生命或者严重影响生存质量的疾病,且尚无有效防治手段或者与现有治疗手段相比有足够证据表明具有明显临床优势的创新药或者改良型新药等,可以申请适用突破性治疗药物程序。治疗严重危及生命且尚无有效治疗手段疾病的药品,药物临床试验已有数据证实疗效并能预测其临床价值的,可以申请附条件批准。多重加快审评程序的综合应用,极大地加快了罕见病药品的上市速度。
3.3 批准情况
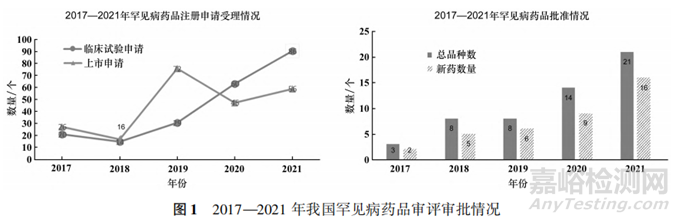
近年来,随着药品审评审批制度的建立健全、审评能力和审评体系现代化建设水平的不断提升,药审中心紧密围绕以患者为中心的监管理念,推进临床急需药物进一步加速上市惠及患者。在罕见病药品审评方面,药审中心通过“快速通道”和临床急需境外新药审评审批工作程序为罕见病药品开辟“绿色通道”,合理配置审评资源,实现药品“急用先行”。通过这些加快措施,2022年获批的进口罕见病药品绝大多数豁免境内临床试验,境内外上市时间差约为3年。2019-2022 年共有64个罕见病品种通过优先审评获批上市,2022 年 22 个罕见病药品中,进口药 10 个、国产仿制药6个、改良型新药1个,覆盖神经、代谢、血液、心血管、皮肤等多种疾病治疗领域,明确包含儿童适应证的药品有 11个。这些药品极大提升了罕见病药品的可及性,如4个进口药品直接填补我国相关疾病治疗领域临床空白,首个仿制药醋酸艾替班特注射液获批后,原研药价格降低了 48%,减轻了罕见病患者的用药负担,见图1。
四、建议
4.1 罕见病药品存在的问题
随着改革的脚步不断前进、政策的红利持续释放,罕见病患者药物可及性得到较大地改善,如佩索利单抗是首个获批的进口1类生物药,企业在国际多中心临床试验结束后同步向中国、美国和欧洲提交上市申请,我国批准时间仅比全球首次批准(美国)晚100 d。但是,与其他药物对比,罕见病及罕见病药品具有与一般医疗产品不同的特点和研发规律[10]:① 从发病规律角度,罕见疾病中很多是严重或危及生命的疾病,许多属于先天遗传性疾病且多始发于儿童期。② 从统计规律角度,罕见疾病的患者群体小,流行病学和疾病自然史等数据有限,医疗信息不充分,可能缺乏公认的疗效评价方法和研究终点;疾病可能存在多种亚型,不同亚型患者的症状、体征、患病率及进展模式等可能各不相同,患者可能呈现较高的异质性,以儿童患者为主的临床研究方法具有特殊性。③从研发成本角度,罕见病药物开展临床研究的机会有限,研发经验较少,成功率低,上市后销售范围有限,投入产出比低。④ 从制度保障角度,罕见病的定义尚无统一规范标准,有关规范性文件列举出的罕见病种类较少,罕见病研究诊疗的前提基础依旧薄弱。同时,罕见病药品加快审批制度虽然已经建立,但在设立专项基金鼓励企业研发、减免罕见病药品生产企业研发费用方面的相关保障措施仍然十分缺位。⑤ 从社会影响角度,罕见病儿童患者居多,且严重危及生命健康,因此罕见病不仅是个人健康问题,而且关系到整个家庭的稳定和发展,关系到社会对儿童群体的关心关爱。这些独特性凸显出我国罕见病药品在研究、生产、监管、使用等环节还存在一些不足:① 罕见病基础研究仍存许多空白,临床资源有限,发病机制不明,② 企业对罕见病药物研发投入不足、风险大、回报少。③ 患者对罕见病药品需求迫切,希望有救命药、便宜药。④ 监管部门要保障患者用药安全、有效、可及,激励企业投入研发生产,促进产业良性循环。
4.2 政策建议
4.2.1 制定罕见病药品认定标准
从部分国家和地区的经验来看,通过立法对罕见病及罕见病药品进行界定,从发病率、患病率、药品可及性等角度对罕见病及罕见病药品进行界定,为企业划定更加精确的发力点,精准地释放制度优势,优化资源配置有力地推动了罕见病的保障机制的发展。
4.2.2 设立专门机构负责罕见病药物管理
目前我国的罕见病认定、罕见病药品的批准、临床使用、医保保障等职能分散在国家卫生健康委员会、国家药品监督管理局、国家医保局等部门,且暂未设立常设工作机构(或机制)。现有的管理方式可以充分发挥各部门的职能优势,提高管理效率,但在强调分工的同时,组织协调成本较高。建议设立专门机构或建立常态化工作机制,对于罕见病及相关药品统筹管理,打通研产用保通道。同时,在统一归口管理的制度下,完善对罕见病药物的研发指导、审评技术标准体系、临床开发资源等配套措施。
4.2.3 丰富针对孤儿药研发的激励政策
如免除监管费用、提供研发资助、提供协议援助、给予市场独占期等。目前我国规定了罕见病药品加快审批、减免税额、纳人医保等制度保障措施,一定程度上较好地激励了企业从事罕见病药品的研发和生产。但对比发达国家或地区监管机构的激励方式,仍存在一定的缺位和缺失。如罕见病药品的市场独占期制度,美国规定了7年,欧盟、日本规定了 10年。专利制度和数据保护制度都属于知识产权保护的范畴,本质上是给予创新者市场独占的奖励。建议从鼓励企业投人罕见病药物研发、稳定企业开发的市场预期角度,参照日本、欧盟做法,规定罕见病药品上市后享有 10 年的市场独占期,并对儿童罕见病药品适当延长,形成罕见病产业的良性循环。2022 年5月发布的《中华人民共和国药品管理法实施条例(修订草案征求意见稿)》对罕见病的市场独占期予以相应规定[11]。
参考文献
[1]逯军,潘翔.浅谈我国罕见病的研究现状与未来[J].中国热带医学,2023.23(2):109-114.
[2] 国家卫生健康委员会,科学技术部,工业和信息化部,等.关于公布第一批罕见病目录的通知[EB/OL].(2018-05-11)2023-11-10].http://www. nhc.gov.cn/yzygj/s7659/201806/393:9a37139c4b458d6e830f40a4bh99.shtml.
[3] 李诗杨,殷彩琴,周楠,等.“互联网+”背景下孤儿药可及性提升策略研究--基于长尾效应[J].卫生经济研究,2022.39(9)49 -52
[4] 国家市场监督管理总局,药品注册管理办法[EB/OL].(2020-01-22)[2023-11-10].http://scjgj.yibin.gov. cn/sy/xxgk/zcwj/202004/20200407 1260862. html.
[5] 张金子,宋晓琳,王泽钊,等.国外孤儿药研发激励机制对我国的伦理启示[J].中国医学伦理学,2022,35(9):971 -977.
[6] 张海琴,李顺平,冯俊超,等.典型国家罕见病医疗保障发展的历史逻辑及相关启示[J].中国医疗保险,2022(5):112-115.
[7] 国家药品监督管理局综合司,中华人民共和国药品管理法实施条例(修订草案征求意见稿)[EB/0L].(2022-05-09)2023-11-10].https://www.nmpa. gov. cn/xxgk/zhqyjzhqyjyp/20220509220456183. html.
[8] 郑建洪,邱春风,张真.基于 ClinicalTnals.sov 的中国罕见病目录临床试验注册现状分析[J],中国新药杂志,2023,32(16):1600-1607.
[9] 周朋,任思宁,刘媛,等.中国已纳人优先审评审批程序的药品注册情况分析[J].中国新药杂志,2024.33(6):534-542.
[10] 王晓建,罕见病精准医疗:呼吁更多行动[J].中国医药导刊,2022.24(1):29-32.
[11] 中华人民共和国药品管理法实施条例(修订草案征求意见稿)[EB/0L].(2022-05-09)[2023-11 -10]. https://www.nmpa.gov.cn/xxgk/zhqyj/zhqyjyp/20220509220456183. html.

来源:中国新药杂志


