您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-08-12 20:00
近期NBCG发布了最新指南:NBCG-MED 2024-1,混合审核在 MDR/IVDR 质量管理体系评估中的应用。下面,本文将对该指南的重要内容进行解读:
1、NBCG-Med
NBCG-Med是根据MDR 2017/745 法规Article 49 和IVDR 2017/746法规Article 45,成立的医疗器械领域的公告机构协调小组。MDR和IVDR下指定的所有公告机构都参与了该小组的工作,并致力于:
a) 允许公告机构就合格评定程序的应用和公告机构的活动分享经验并交换意见
b) 促进公告机构及其流程之间的一致性
c) 起草技术建议,并就与合格评定和公告机构活动有关的事项达成共识
d) 应委员会和MDCG的要求,就医疗器械立法及其实施向委员会和MDCG提供建议
e) NBCG-Med 有一个技术小组,即公告机构技术小组,分为医疗器械主题 (NBTG-MD) 部分和体外诊断主题 (NBTG-IVD) 部分。
2、NBCG-MED 2024-1指南解读
► 开发和采用说明
本文件最初由公告机构制定并通过,作为 Team-NB 立场文件“公告机构关于混合审核在 MDR/IVDR 下质量管理体系评估中的应用”。
在此版本中,该文件已进行了更新,考虑了 MDCG公告机构监督小组的意见,并被采纳为 NBCG-Med 的立场。这是该文件作为NBCG-MED立场通过后的第一个版本。
►背景
传统意义上,质量管理体系(QMS)审核是在现场进行的。然而,在全球疫情大流行期间,公告机构实施了一些程序,根据适用的要求和指南,利用信息和通信技术 (ICT) 应用替代方法,并与适用的要求和指南保持一致(例如 MDCG 2020-44 和 IAF MD 4)。
本文件是根据 MDCG 的建议制定的,旨在进一步阐述“混合审核”的操作要素,包括确定在受审核方场所进行审核的方面,如 MDCG 2022-17 所述。本文件代表了公告机构对基于信息和通信技术(ICT)的审核方法("混合审核")的集体立场,特别是在根据 MDR/IVDR 进行质量管理体系审核时需要考虑的方面。
► 混合审核的法规背景
作为初次审核和监督审核的一部分,公告机构必须对制造商的质量管理体系进行现场审核。关于初次审核,MDR/IVDR附件IX第2.3节规定:评估程序应包括对制造商的场所进行审核,并在适当的情况下对制造商的供应商和/或分包商的场所进行审核,以核实制造和其他相关过程。
关于监督审核,MDR/IVDR Annex IX section 3.3规定:NB 应定期(至少每 12 个月一次)进行适当的审核和评估,以确保有关制造商实施经批准的质量管理体系和上市后监督计划。这些审核和评估应包括对制造商的场所进行审核,并酌情对制造商的供应商和/或分包商进行审核。
根据这些要求,如果使用基于ICT的替代方法对MDR/IVDR进行质量管理体系审核,则这些审核的至少一部分必须在现场进行,以涵盖制造和其他相关过程,即审核必须是MDCG 2022-17中定义的混合审核:
“混合审核”应理解为在制造商或其供应商和/或分包商的场所进行的审核,至少有一名审核员在场,审核小组的其他成员从其他地方使用信息和通信技术(ICT)参与。
由具有适当资格的工作人员进行的这种混合审核将满足上述MDR/IVDR的现场审核要求。
根据疫情期间获得的经验,如果规划得当,混合审核是有效的,与完全现场审核相比,具有以下优势:
a) 根据公告机构的估计,与现场审核相比,混合审核可以节省高达25%的审核能力,从而可以将节省的能力重新用于进行额外的MDR/IVDR审核,以帮助从指令进行整体MDR/IVDR过渡,从而更有效地利用审核员的能力;
b) 减少在旅行和住宿上花费的时间和精力,从而减少旅行限制;
c) 降低前往高风险地区(例如政治动荡、大流行、自然灾害)的风险;
d) 降低审核员职业倦怠的风险;
e) 混合审核更具可持续性,并减少了审核对环境的影响;
f) 混合审核促进了包容性
► 混合审核要求
制造商质量管理体系的某些方面可通过 ICT 进行有效审核,但某些方面应在混合审核的现场部分进行审核。使用信息和通信技术可有效审核的方面以及现场审核部分应审核的方面包括(但不限于)下表所列的内容。
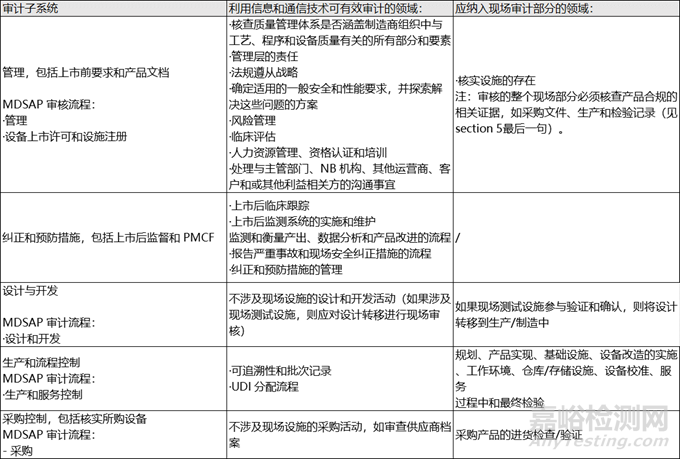
► 审核团队资质
在MDR/IVDR混合审核的背景下,审核团队必须满足MDR/IVDR附件VII第3.2.6节中规定的与现场审核员相关的资格标准。
执行混合审核的现场部分的现场审核员应符合适用于审核范围内的流程的 MDT/IVT 代码,这些流程在审核设施中实际发生,并且作为现场审核员对设备和设备相关技术有足够的了解,视情况而定与审核活动相符。在实际到被审核设施的审核员无法满足所有必要资格的情况下,具有适当资格的其他审核团队成员必须通过ICT同时支持审核。在这种情况下,审核期限应考虑审核小组成员审查相关过程所需的额外时间。
► 审核计划和持续时间
作为审核计划的一部分,公告机构必须考虑制造商的能力,以及支持混合审核(IT系统、纸质与电子QMS文档和记录等)的适用性。应根据IAF MD 59和IAF MD 910中规定的原则确定总审核期限。
根据GHTF/SG4/N30,大约20-30%的审核持续时间分配给生产和服务控制子系统的审核。因此,必须将至少 25% 的混合审核持续时间分配给审核的现场部分。审核的现场部分应适当增加,以反映审核持续时间计算中适用的增加系数,这些系数适用于在被审核设施实际发生的制造商生产活动。
在有正当理由的情况下,可以减少现场审核的部分。示例包括(但不限于):
a) 没有实际发生生产活动的设施,需要审核员在现场进行审查,例如,仅生产作为医疗设备生产软件(SaMD)的设施,其中生产活动仅使用简单的流程或所有生产活动都完全外包(“虚拟制造商”),并且没有实际处理任何产品;
b) 仅进行行政活动的设施,例如人力资源管理、采购或其他没有实际产品处理的管理流程。
然而,在这些情况下,审核的现场部分必须验证设施的存在,并在相关情况下验证产品合规性的证据,例如采购文件、生产和检查记录。

来源:Internet


