您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-11-08 18:17
1、 三起毒素门事件给制药行业带来的警醒和反思
2007年6月6日,EMA发现部分批次的抗爱滋病药物Viracept,即奈非那韦甲磺酸盐(1)中含有高剂量的基因毒性杂质(genotoxic impurities,GTI)甲磺酸乙酯(2)(含量超过1.1×10-3),决定召回由罗氏制药生产的1(化合物结构见图1)。
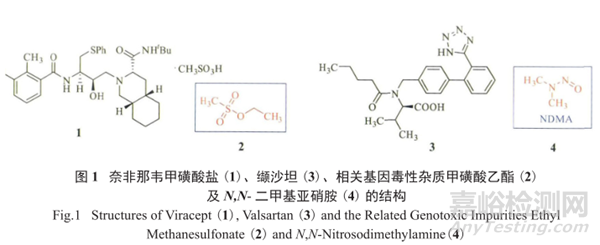
2 是潜在的致癌物质,在药物剂量为2.5g/d的情况下,高达2.75mg的2可能被患者摄入体内,进而影响到患者的身体健康。
无独有偶,2018年7月14日,华海制药生产的高血压用药缬沙坦(valsartan,3)由于检测出含有微量(含量大于0.3×10-6,即0.3ppm)的基因毒性杂质N, N-二甲基亚硝胺(NDMA,4),3及其相关制剂也被宣布从欧洲、美国和中国市场上召回 (化合物结构见图1)。
不幸的是,2018年8月20日,在印度Torrent制药生产的缬沙坦片剂中也检测出含有 NDMA,该公司也从美国市场上自愿召回了14批次的相关药品。
这三起毒素门事件不仅给病人带来了安全隐患和健康问题,而且也给相关药企带来了不可估量的经济损失,给整个制药行业敲响了安全生产的警钟。
2、 基因毒性杂质的基本概念与控制原则
基因毒性杂质是指能直接或间接损害DNA、引起基因突变或致癌的一类物质。其对 DNA的损害作用包括染色体断裂、DNA重组及复制过程中共价键的插入和修饰,也包括在细胞水平上产生基因毒性物质而产生的突变。
随着科学的进步和药政监管的日趋完善,2006 年欧洲EMA、美国医药研究与制造商协会(PhRMA),2008年美国FDA先后颁发了关于基因毒性杂质限度指南 (Guideline on the Limits of Genotoxic Impurities),和一套基因毒性杂质分类、界定、检测和风险评估的程序 ;
2014年人用药品注册技术要求国际协调会议(ICH)又进一步修订了同时满足欧美的关于基因毒性杂质的指南ICH M7,这样对药品进行充分的基因毒性杂质研究也成为药品能否获批,能否上市的关键因素之一。
基因毒性杂质从何而来?哪些化学物质和官能团具有潜在的基因毒性?这类物质有没有方便判断的警示结构(structural alerts for genotoxicity) ?
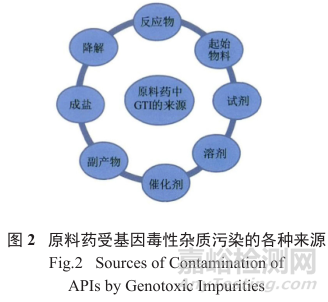
显然,基因毒性杂质是从化学试剂、化学合成与反应作用而来的,涉及到合成工艺流程的方方面面以及随后的药品的稳定性和可能的降解,是一个极其复杂的过程问题( 图2)。
在基因毒性的控制过程中,一个重要的概念就是毒理学关注阈值(threshold of toxicologicalconcern,TTC),其限度(1.5µg/d) 作为基因毒性杂质的可接受限度,低于该限度,则不能观察到显著的毒理作用。具体定义是:在人的一生(70岁)中,每天摄入1.5µg的基因毒性杂质,其致癌的风险是可以接受的。
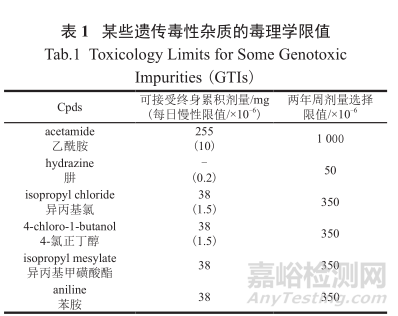
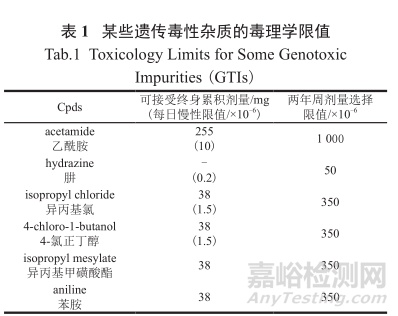
表1中列举了一些常见的GTI的毒理学限值。例如成年正常人体对乙酰胺可接受的每日的慢性限制是摄入量小于10×10-6(即10ppm),而可接受的终生累积剂量为255 mg。
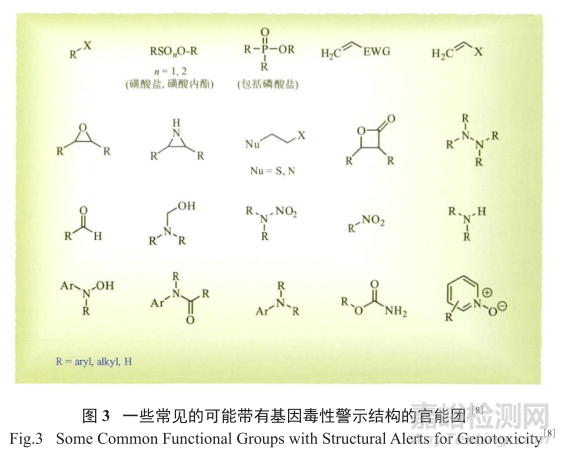
3、 可能具有基因毒性的警示结构
图3列出了一些常见的带有基因毒性警示结构的官能团,也称为Ashby和Tennant简化系统,这些官能团涉及到的与DNA的反应众所周知。尽管这些结构并不详尽,但却是进行基因毒性杂质结构评估的基础。
其他的结构评估商业软件有DEREK和Mcase。当基因毒性化合物,如环氧化物或芳香胺作为反应物和试剂参与反应制备原料药时,极有可能给最终的活性药物成分 (API)带来基因毒性杂质污染,因此对GTI的严格检测、控制和预防在工艺化学过程中是必须的。
表2为可能带有基因毒性警示结构的20类有机化合物。根据PhRMA的模型,由化学反应带来的基因毒性杂质分为五类:
①已知具有遗传毒性和致癌作用的杂质 ;
②已知具有遗传毒性,但潜在致癌性未知的杂质 ;
③具有警示结构,但与最后的API结构无关的杂质 ;
④具有与API有关的警示结构的杂质 ;
⑤无警示结构或有足够证据证明无遗传毒性的杂质。
对第一类杂质要避免,甚至必须更改合成工艺 ;
对第二类、第三类杂质要进行风险评估和必要的掺杂(spiking,又译为加标),和清除(purging)操作,跟踪、推断并使用TTC阈值调节机制来进行质量控制 ;
第四类和第五类杂质则可以按一般杂质来处理,即含量要低于0.1%。
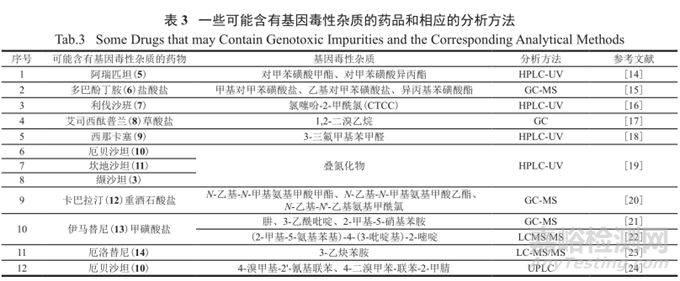
从基因毒性杂质研究指南的出台和认知接受过程(2000—2008)中可以看出,制药工业界的认知和了解是不断加深的,2010年OPRD(Organic Process Research & Development)期刊还专门组织发表了关于基因毒性研究的专刊,对特定上市药物的基因毒性物质的研究也有文献发表,表3和图4罗列了近年来一些可能含有基因毒性杂质的常用药品,及笔者判断的可能来源和相应的分析方法。
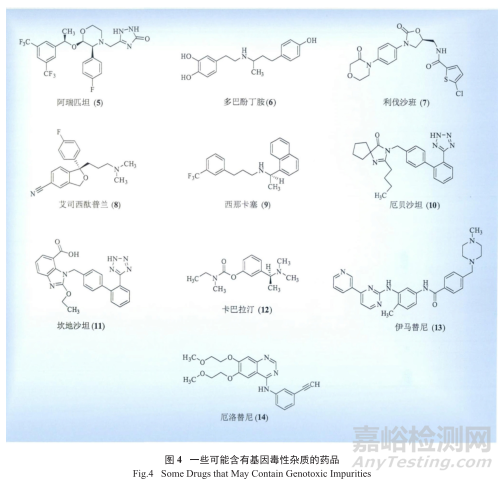
图5是在原料药中确定、控制和测定基因毒性杂质的流程图(或称之为决策树)。从图中不难发现该流程涉及到整个合成过程,尤其是下游的工艺化学,而且和毒理的评估、基因毒性杂质的鉴定和过程的控制方案,以及分析方法的选择和使用紧密相关,是一个复杂细致和逻辑分析的过程。
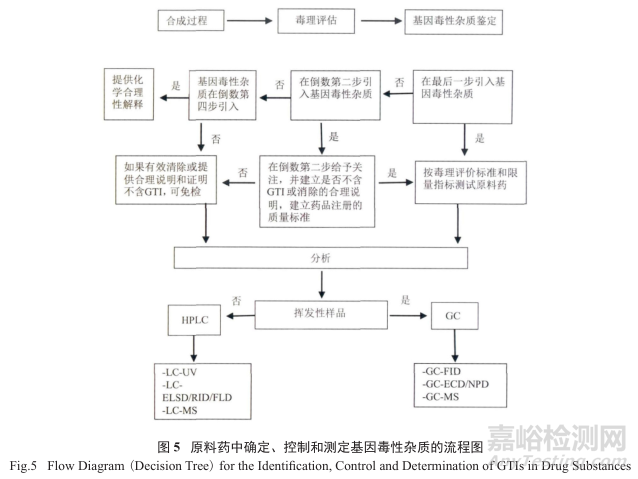
值得强调的是 :如果基因毒性杂质在最后一步中引入,则必须根椐毒理学的评估报告,设立其限制标准(极限测试 ) ;如果基因毒性杂质是在远离最后一步反应(在4步以上)引入的,则可以通过证明后续化学工艺中每步环节的合理性,证明可有效地清除GTI来免除控制(免检GTI)。
综上所述,API中基因毒性杂质的来源是多方面的,从原料和反应物的化学作用形成的、并且和最终产物的构造与副反应产物结构相似相关是较为容易鉴别的,而从催化剂、试剂(不参与API构建)、反应溶剂以及产物降解带来的基因毒性杂质有时则不易鉴定。
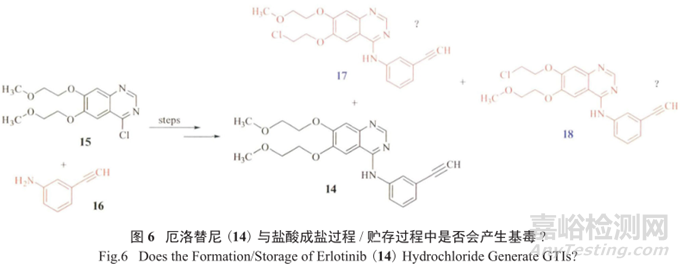
值得一提的是,要想对基因毒性杂质的产生以及可能的结构做准确的判断,需要有很好的专业知识和理论依据作基础,尤其需要对一些反应细节和工艺流程进行深思熟虑。例如表3中对厄洛替尼(14)的基因毒性杂质检测只检3-乙炔苯胺(16)是不够的 ;
厄洛替尼在成为盐酸盐的过程中是否会产生基因毒性杂质17和18,以及盐酸厄洛替尼在贮存过程中会不会降解产生17和18,都是值得关注的(图6)。
4、 基因毒性杂质的“避免 - 控制 - 清除”(avoid-control-purge) 策略
在药物研发和原料药工艺控制上的应用,在工艺研究中采用“避免- 控制 -清除(ACP)”的策略(表4、表5) 能够最大限度减少基因毒性杂质对最终产品即原料药API 的影响。
例如美国礼来制药(Lilly)采用的GTI(或PGI,potentially genotoxic impurities,GTI和PGI这两个相关的名词在许多场合往往交替使用)清除策略就很好地体现了上述原则:
1) 至少把GTI(或PGI)的生成放在离最终产物4步以外,并且在每一步分析工艺能否清除PGI的可能性 ;
2)对上述工艺进行耐受性研究和处理,在分离纯化之前,通过添加(spiking) 额外的 GTI(或PGI) 来检验纯化的强度。
例如,有时加入高达5%的GTI,然后观察每步反应条件对该杂质的清除能力,目的是使工艺集中于起始原料或中间体的上游控制。这种方法的优点是允许控制限度设定在较高水平或可接受的程度,同时也可以使用不太复杂和灵敏度要求不是很高的分析方法 ;
3) 在倒数第二步产物检查PGI的浓度含量,设置极限测试,验证结果,这样最终产品就有可能免检GTI。


对潜在遗传毒性杂质进行风险评估的策略就是利用质量源于设计(quality by design,QbD) 提供更详尽的对工艺和分析的理解(图7),最终确定杂质产生的临界区域。
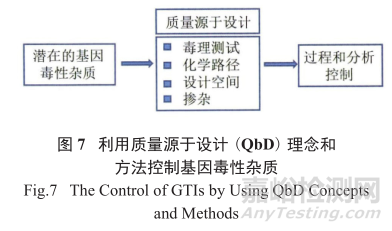
具体在生产工艺开发中,结合产品质量属性,对相关工艺步骤进行筛选优化,通过设置工艺参数控制范围和建立中间体验收标准,保证工艺稳健性和产品质量一致性。
在下游工艺的开发阶段,结合工艺相关基因杂质的去除效果和残留量标准,对纯化工艺关键参数进行验证。通过识别确定影响最大的风险和过程参数,可以增强对产品质量和过程的控制,从而减少这些杂质的潜在影响。
这种方法要求在必要时使用毒理学测试,尽可能用化学结果来论证,用开发设计空间的多变量分析,以及使用数据来支持规范。通过强大的分析方法的支持,尤其是低水平检测方法的开发,这一策略不仅有助于开发强大的API工艺,而且还可以识别并减小研究人员高度关注的一类杂质的风险。2012年,Madasu 等报道了商业化切实可行的氯沙坦钾盐(25)的五步合成方法,以55.5%的收率和99.9%的纯度制备了 API(图8)。
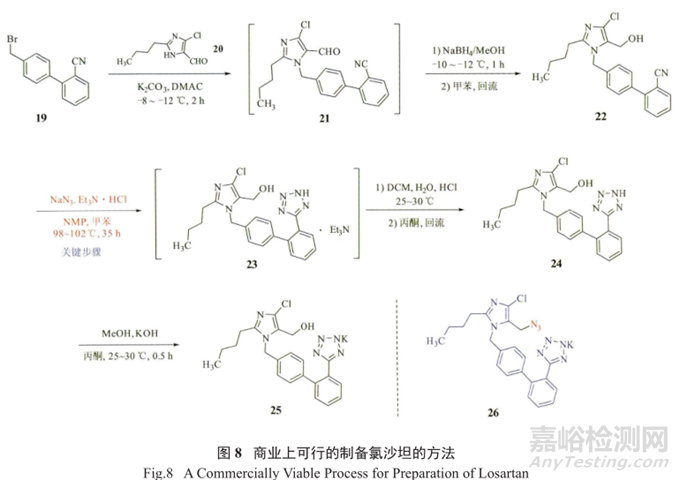
其亮点是在四氮唑的合成中采用了叠氮钠,使用了N-甲基吡咯烷酮(NMP)/甲苯作为反应溶剂进行 [3+2] 偶极加成。美中不足的是反应温度较高、反应时间长达35h ;
但整个工艺过程合理利用了叠缩工艺,避免了不必要的分离纯化,经济可行且满足了法规要求 ;而且从氰化物到四氮唑的反应条件温和可控,也克服了叠氮酸副产物的易爆问题。
值得强调的是,在这个工艺过程中,并没有检测到基因毒性杂质NDMA( 使用的偶极溶剂是NMP,没有使用DMF),但发现含有0.02%的叠氮化物26,该化合物在处理后可以清除。
缬沙坦(3)原料药的生产工艺中四氮唑杂环的形成是工艺的重点和难点。原有工艺存在转化率低、 “三废”产生量大、异构体杂质水平波动等问题。
研究人员以价廉易得的叠氮钠代替昂贵的叠氮三烃基锡,但在芳香溶剂中存在溶解度不佳的情况,故采用溶剂DMF,这样溶解度提高,反应速率加快,反应温度降低,反应也更趋于安全 ;
文献报道,改进后的工艺(图9),转化率提高约30%,“三废”产生量降低约30%,异构体杂质也得到了有效控制。
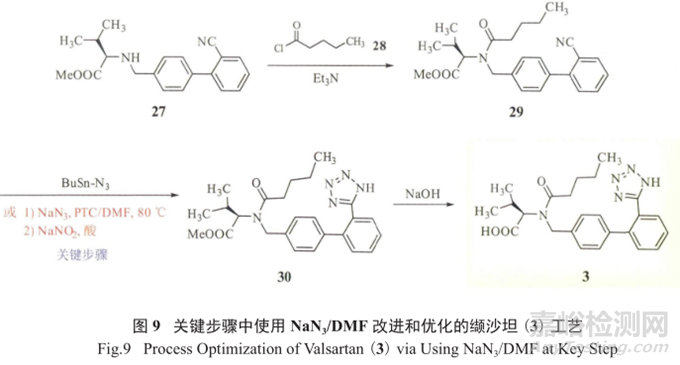
但改用DMF(溶剂中可能含有二甲胺杂质)以及随后的用亚硝酸淬灭、调酸处理,可能引入基因毒性杂质NDMA。
如何既保留采取DMF/叠氮钠有效快速安全地制备四氮唑的优点,同时又克服或清除 NDMA带来的危害呢?显然最合理的策略就是将产生NDMA的化学步骤放在整个合成序列的上游部分,给下游的后处理和控制带来更多的机会。
令人欣喜的是,德国的Ackermann和日本的Seki分别综述了利用Ru催化进行C-H活化,不经过经典的Suzuki偶联反应,直接进行C-H芳基化,非常合理有效地制备了一系列沙坦类高血压用药(图10)。
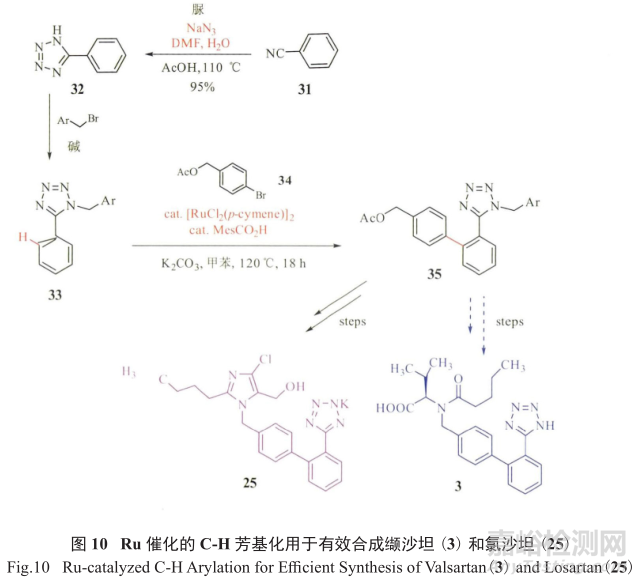
将可能产生GTI的步骤前移至工艺流程的上游部分,将有毒物质和试剂尽量远离最终的API。另一例子是辉瑞制药推出的第一个治疗勃起功能障碍(ED)的口服药物西地那非(又称“伟哥”,38) 的合成工艺(图11)。
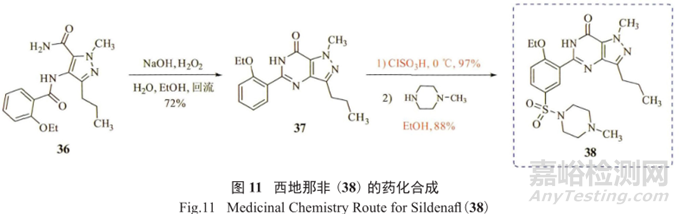
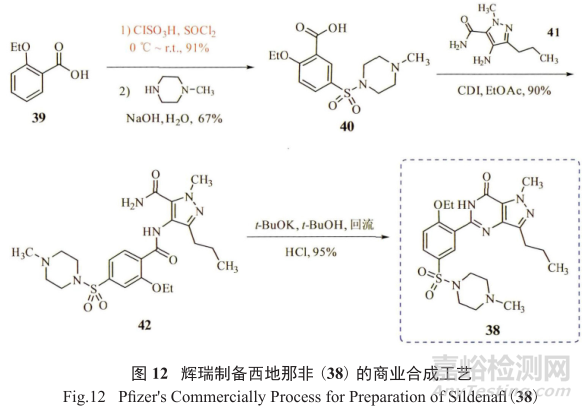
原来的合成方法将磺酰胺化放在倒数第二步,给潜在基因毒性杂质的产生(硫酸单乙酯和硫酸二乙酯)、防范和控制带来了不小的挑战,为此,辉瑞制药的工艺化学家重新设计了一套符合绿色化学原理的商业化合成工艺路线(图12),将磺酰胺化反应放在整个反应过程的第一步,取得了很好的效果。
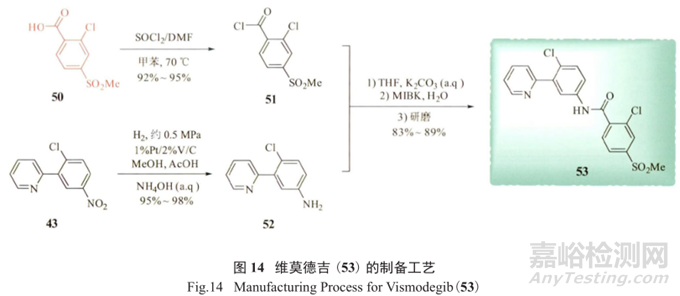
罗氏制药在抗肿瘤药维莫德吉 (vismodegib,53) 研发合成工艺过程中,发现存在主要潜在基因毒性杂质50 ;同时在经氢化反应制备关键中间体52的过程中,发现不彻底或过度的氢化都会带来具有警示结构的新杂质羟胺44和芳香胺48、49(图13),给质量控制带来了不小的挑战。
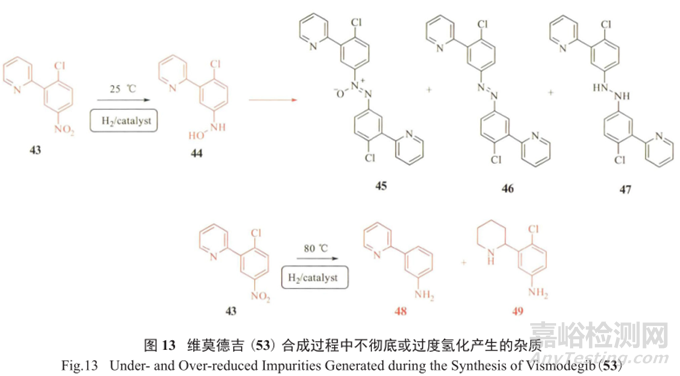
可通过优化氢化反应条件来严格把控潜在基因毒性中间体50。根据指导原则,每人每天的遗传毒性杂质摄入量不能超过1.5µg,即毒理学担忧阈值为1.5µg/d。
也就是说,从维莫德吉每天每人给药量150mg来算,该药物中含有的这些遗传毒性杂质含量需要在10-5以下。在维莫德吉的合成中,合成工艺研究人员通过原料中的杂质限度控制以及改变反应条件,最终将维莫德吉原料药中的潜在基因毒性杂质50 的含量控制在10-6以内(图14)。
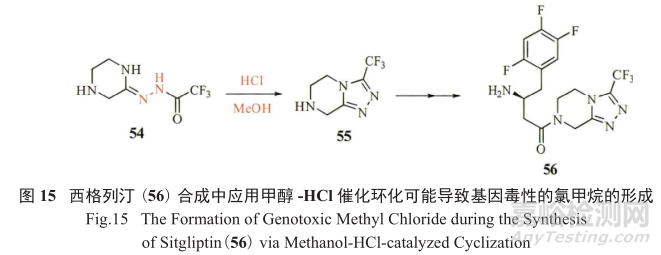
一般来说,药物的杂质多是在合成过程中生成的副产物,其结构往往和目标分子或中间体结构类似,但不可否认,有时产生的GTI可能跟目标产物和中间体都没有关联。
例如在抗糖尿病药物西格列汀(56)的制备过程中(图15),第一步甲醇和HCl的组合就有可能产生基因毒性杂质氯甲烷;在一些抗肿瘤药物如伊马替尼成盐的过程中,如果使用了醇类溶剂,则可能产生相对应的甲磺酸酯GTI;
当乙腈水溶液在酸性条件下长时间加热时,也会生成基因毒性物质乙酰胺,尽管其水溶性很好,易于排除。药物成盐是药物最广泛的存在形式,它不仅改善了药物的理化性质,提高了药物的溶解度和生物利用度,而且还使药物的晶型可控,提高了药物的稳定性。
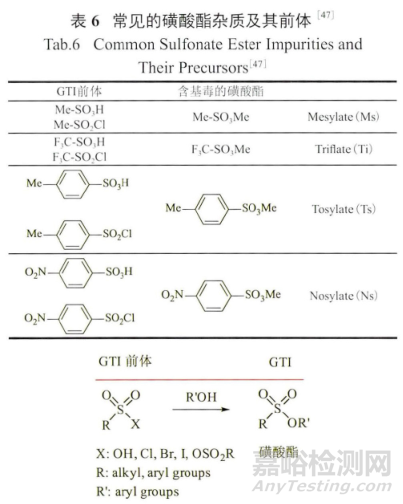
据统计,在20种常见的用于制备盐的酸中,甲磺酸的比例高达3.2%,同时磺酸(包括三氟甲磺酸、对甲苯磺酸和对硝基苯磺酸)酯(表6)作为好的离去基团和催化剂常参与环合反应,因此如果这一类酸存在,含有被甲磺酸酯和其他的磺酸酯污染的API和最终药品的可能性很大;所以最好能避免在醇类溶剂中成盐,但许多药物的最佳晶型都是在醇类溶剂中结晶出来的,而且有些反应的溶剂就是醇,这就给药品质量控制带来了极大的挑战。
如何控制磺酸盐的生成呢?一般的操作规律如下 :
①避免使用过量的磺酸 ;
②控制和严格把关磺酸的纯度 ;
③如果需要过量的磺酸,则需要很好地控制用量,避免大过量 ;
④在成盐和分离过程中,尽量采用切实可行的低温条件 ;
⑤避免磺酸和醇过,早混和贮存在一起,减少磺酸与醇接触的时间,以防磺化反应的发生 ;
⑥利用位阻大的醇,以减少酯化的可能性,如i-PrOH<EtOH<MeOH ;
⑦如果可能的话,在成盐和分离过程中,添加少量水或用醇/水体系,将酯化的平衡移向醇和水的方向。有水存在时,在醇中生成甲磺酸酯的速度变缓。
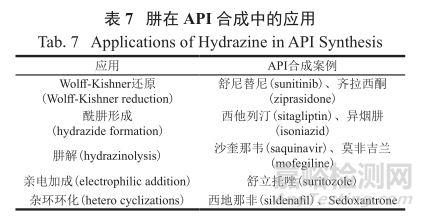
值得注意的是,有时采用基因毒性物质作为反应试剂和原料是不可避免的,例如使用肼和肼的衍生物(表7)。抗肿瘤药物舒尼替尼(59)、抗精神病药物齐拉西酮(60)以及抗糖尿病药物西格列汀(56)等都使用肼作为还原剂或是作为合成关键杂环的原料(图 16),一条不成文的规矩是 :尽量把肼或肼的衍生物的化学合成放在合成工艺的上游部分,这样就能提供更多的机会将相关的基因毒性杂质控制在可控的TTC值以下。

在体外试验中发现,对甲苯磺酸异丙酯比其他磺酸酯的致诱变性更强。葛兰素史克 (GSK)在地格列汀(denagliptin) 甲苯磺酸盐(62)项目上就很好地利用了“控制-清除”策略来达到安全生产API 的目的:
1) 在水存在条件下脱保护,尽量减少任何生成酯的可能 ;
2) 发现对甲苯磺酸盐粗品中含有的对甲苯磺酸异丙酯,利用重结晶 (72 ℃,1h)水解对甲苯磺酸异丙基酯(图17)。

最近,有文献报道了一种新型非麻醉性镇痛药盐酸奈福泮(68)的公斤级合成工艺(图 18)。
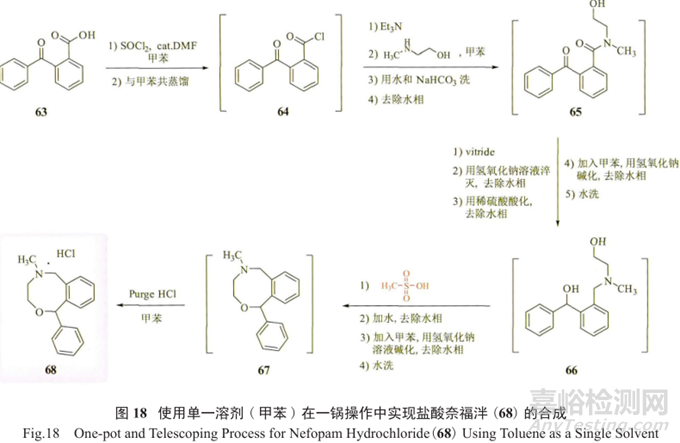
本品与阿片类药物和其他镇痛药相比具有非阿片(非麻醉)和非甾体作用,副作用较少,因此作为镇痛剂在大多数欧洲国家广泛应用。该工艺使用单一溶剂 (甲苯)在一锅中制备盐酸奈福泮API,总收率良好(≥79%),纯度≥99.9%。
一锅5步合成工艺包括 :由苯甲酰苯甲酸(63)形成酰氯64,然后经酰胺化、还原、环化,最终成盐酸盐68。
主要优点有 :
①使用单一溶剂,
②每步转化率均大于90%,
③成本效益高且操作便捷,
④经一锅操作,总收率(≥79%)提高。
该工艺有如下特点 :
合理选择了高效的还原试剂双二氢铝钠(vitride,Red-Al),一步有效还原了酰胺和酮成为胺和二级醇;同时通过筛选,采用甲磺酸/甲苯条件,以“一锅煮”的操作方式有效成环制备了API。
文献称该工艺能够合理地避免基因毒性杂质的形成。但从最终纯化68时采用了甲醇的情况以及甲磺酸的使用来看,若有潜在的基因毒性杂质69~73(图19)的形成,则经酯交换反应,极有可能带来甲磺酸酯这样的GTI(注 :该文献作者发现未知杂质经甲醇、乙醇或甲醇/水纯化处理后的含量在0.01%~0.2%之间)。

葛兰素史克(GSK)和阿斯利康 (AstraZeneca)的Elder和Teasdale对OPRD在过去10年间(2001—2010) 发表的300多篇研究论文的调查统计也高度概括和总结了当前 API的合成策略。
它再次肯定了制药行业的普遍认知,即在复杂的、多步的药物合成中不使用有高反应活性、有潜在诱变性的中间体是不现实的,而完全“避免”使用这些中间体则反映了对现代合成化学认识的缺乏。
根据这项调查还可得出结论 :合成一种API所需的平均步数为6步,一条合成路线的平均活性中间体数目为4;还注意到,在API合成的后期阶段(即最后4个阶段)中通常主要使用4种类型的反应性中间体 :烷基卤化物、酰氯、芳族胺及迈克尔受体。
此外,无论选择哪种途径,反应中间体的数量仍然大致相似,这强烈挑战了“避免”永远是可行的选择的观点。上述结论也再次证明了控制,而不是回避,是绝大多数情况下最合适的策略。
很多抗肿瘤药物都有芳香胺的结构,其中间体大多也具有芳香胺、硝基、芳香酰胺结构,按照基因毒性警示结构,这些中间体都具有基因毒性。
但是基因毒性杂质的 TTC 值是基于正常人生命周期来计算的。ICH M7明确规定 :“该指南不适用于晚期癌症病症指征所用原料药和制剂”。
例如沙利度胺(thalidomide,74,图20) 曾是一种镇静和止吐药,最初于20世纪 50年代在临床中用于治疗孕吐,但由于超过10000 名婴儿出生时患有严重的先天缺陷而被禁用 ;
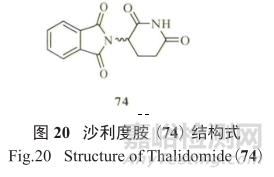
但在1998年,沙利度胺被批准用于治疗多发性骨髓瘤 ;最近令人信服的支持沙利度胺的结构证据是,二羟基酞嗪的氧化代谢产物导致了 DNA 损伤。
此外,可能在某些情况下,药物本身在治疗浓度下就具有基因毒性,会使癌症风险增加。在这种情况下,暴露在具有诱变性杂质下,不会显著增加原料药的癌症风险。
因此,杂质可以被控制在非基因毒性杂质的可接受水平。这些具有警示结构的中间体在没有明确其不是基因毒性杂质时,应该作为潜在基因毒性杂质。
如果在生产工艺开发过程中,精制后没有检测到这种杂质,则不需要列入质量标准中 ;如果有检测到,则必须对其进行合理控制,甚至需要进行风险评估。
5、 结语与展望
近十年来,在药物研发和原料药的生产过程中,药企和各国药监部门对基因毒性杂质的认识和理解、控制和清除有了较为深刻的变化,发现并解决了一些原料药中的GTI 问题,提出并较好实施了“避免 - 控制 - 清除”策略。
在新药研发过程中,及时发现、认真对待可能出现的基因毒性杂质,通过评估风险、采用科学合理的方法来优化合成工艺是必须遵守的基本原则 :
第一,开发并使用新的合成方法和途径,尤其是开发出新的替代试剂,将GTI的前体试剂排除在外,采用新反应媒介,避免使用或替代有基因毒性或可能致癌的溶剂 ;
第二,对反应过程和反应机理有深刻的理解,对产生潜在GTI的步骤可通过优化调整工艺化学和物理参数将GTI控制在合理的区间 ;
第三,识别和判断相关工艺质量是否有GTI,并加以控制使其在TTC阈值以下 ;
第四,建立灵敏有效的分析方法对工艺过程进行跟踪检测 ;
第五,针对特殊的GTI,发展新颖有效的API纯化技术。
不可否认,防控基因毒性杂质如“鸡蛋里挑骨头”一样,是一件富有挑战性的工作,它涉及到工艺化学、毒理学数据采集和处理评估以及化学分析方法的建立和使用。
采用“避免 - 控制 - 消除”的策略,对反应的过程包括可能的副反应要有深刻的理解,对溶剂尤其是醇类物质在成盐过程和清洁过程的质量体系的管控也至关重要。
对药物研发中基因毒性物质的控制既要高瞻远瞩(从反应机理层次上洞察问题),又要防微杜渐(在细节上有管控措施),做到未雨绸缪。
笔者相信,随着对药物研发中的质量控制和GTI的严控的认识和经验的不断丰富,一定会有更多优质的药品造福百姓。
来源:《中国医药工业杂志》
原标题:《药物研发中基因毒性杂质的控制策略与方法探索进展》
作者:张霁 ,张英俊 ,聂飚

来源:Internet


