您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2018-10-15 11:03
为了微观模拟体系能够反映宏观实验现象, 需要通过周期性边界条件对模拟对象体系进行周期性复制, 以避免在实际中并不存在的边缘效应(edge effects)。原则上,对于任何分子体系的理论研究都需要求解含时薛定谔方程。但在实际工作中,更关注的是原子核的运动轨迹,这样的轨迹可以利用波恩-奥本海默近似(Born-Oppenheimer approximation),通过求解经典力学运动方程获得。Alder和Wainwright曾表示计算机模拟实验会成为联系宏观实验现象和微观本质的重要桥梁,在他们首次进行分子动力学模拟实验之后10 年,法国物理学家Verlet提出了一套牛顿运动方程的积分算法,与此同时提出的还有另一套产生和记录成对近邻原子的算法,从而大大简化了原子间相互作用的计算。这两种算法至今仍以一些变形的形式在实践中被广泛应用[1,2]。
过去几十年开发了多种原子级模拟方法,包括晶格静力学、晶格动力学、蒙特卡罗和分子动力学等。其中,分子动力学特别适用于塑性变形的研究,它通过一些规定的原子间相互作用势函数的原子相互作用系统的牛顿方程的解,研究变形过程中的实时行为,并包括晶格的非简谐性、内应力的高度不均匀,以及系统的瞬态响应等方面的影响。
分子动力学主要依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量和其他宏观性质。它对原子核和电子构成的多体系统,求解运动方程,是一种能够解决大量原子组成的系统动力学问题的计算方法,不仅可以直接模拟物质的宏观演变特性,得出与试验结果相符合或相近的计算结果,还可以提供微观结构、粒子运动以及它们和宏观性质关系的明确图像,从而为新的理论和概念的发展提供有力的技术支撑。
分子动力学的对象是一个粒子系统,系统中的原子间的相互作用用势函数来描述,因此,正确选择势函数的类型及其参数,对于模拟的结果优劣具有重要作用。势能函数在大多数情况将描述分子的几何形变最大程度地简化为仅仅使用简谐项和三角函数来实现;而非键合原子之间的相互作用,则只采用库仑相互作用和Lennard-Jones 势相结合来描述。其中,对于原子间相互作用力的描述通常是经验或半经验的,这样虽然能够提高计算效率,但无法完全揭示电子键合的多体性质,尤其对于缺陷附近与自身结构和化学性有关的复杂自洽变分函数。Daw和Baskws的EAM(Embedded-atom model)势函数在某种程度上融合了电子键合的多体性质,将系统的总势能表示为:
式中:Fi是原子i的嵌入能函数;ρi是除第i个原子以外所有原子在i处产生的电子云密度之和;Φij是第i个原子与第j个原子之间的对势作用函数;rij是第i个原子与第j个原子之间的距离[1]。
势函数的可靠性主要取决于力场参数的准确性,而力场参数可以通过拟合实验观测数据和量子力学从头算数据得到。目前在生物大分子体系模拟中使用最为广泛的分子力场是CHARMM力场和AMBER力场,是早期研究生物大分子的分子力场。现有的力场参数仍在不断优化之中,并且涵盖的分子类型也在不断扩大。粗粒化(coarse-grained)模型在计算生物物理研究中越来越引起人们的关注,由于在该模型中定义了粗粒化粒子,对应于全原子模型中的若干原子或原子基团甚至分子,减少了体系中的粒子数,使得模拟的时间和空间尺度得以大幅度提高,但同时也将丢失原子细节信息。基于这种模型的分子动力学模拟适合于研究缓慢的生物现象或者依赖于大组装体的生物现象。
设计一个基本力场的根本原则就是要单位时间步长(time step)内计算能量的开销最小化,从而实现模拟尺度的最大化。这一点对于全原子力场尤为重要, 即便对于所谓的粗粒化模型也是一样。特别地,如果想要进行微秒甚至毫秒级时间尺度的模拟,这一原则极其重要。
图1显示了分子动力学的时间和空间尺寸的反比关系,图中从左至右依次为:(1)水,细胞的基本成分;(2)牛胰蛋白酶抑制体,一种酶,其“呼吸”行为可以在毫秒级时间尺度上进行考察;(3)核糖体,一个复杂的生物器件,可以对基因信息进行解码并生产蛋白质;(4)紫细菌光合膜片段,拥有2500 万个原子,图中显示的是嵌在磷脂双分子层上的捕光复合物及光化学反应中心[2]。

随着计算机处理器速度的快速增长以及大规模并行计算架构的发展,大规模并行化或专用的架构技术与可扩展分子动力学程序的结合,计算机模拟包含从位错到基于晶界的变形机制的整个晶粒尺寸范围,为探索材料体系的研究前沿领域开辟了新的途径。
例如,William Gonçalves等人通过选用Wolf BKS(van Beest, Kramer and van Santen)势函数来描述原子之间的相互作用力,使用大规模原子/分子并行模拟器LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)对二氧化硅气凝胶的弹性和强度进行了分子动力学方面的研究,他们使用velocity-Verlet算法和1.0 fs时间步长,并且在三个方向上均使用周期性边界条件[3]。
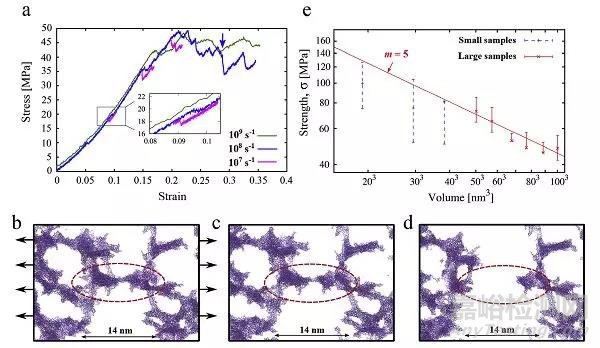
图2为模拟的超过7000000个原子的大体积样品3D示意图,以及20nm厚的样品切片和局部放大图(蓝色为氧原子,红色为硅原子),图3(a)是对803nm3气凝胶样品进行单轴拉伸试验,以获得300K的应力应变曲线,(b-d)是典型韧性断裂图像,(e)抗拉强度与样品体积的对数关系。他们分析认为,为了确保正确评估弹性等机械性能,模拟样品的尺寸至少为孔径的8倍,同时,表面积极高的二氧化硅气凝胶需要相对低的应变率以确保准静态条件。

一般地,临界晶粒尺寸dc约为20-30nm,对于晶粒尺寸(50-100nm)较大的变形主要由位错决定;晶粒尺寸小于30nm时主要由GB变形过程主导,降低晶粒尺寸则会导致强度和流动应力减小,即出现“反Hall-Petch效应”。然而,在fcc和bcc金属中用于模拟GBs的多体和对电位之间的广泛比较揭示了这些不同力描述预测的行为中几乎没有质量差异,这表明多体效应可能不会主导GB行为。
B_ejaud, J. Durinck等人利用分子动力学模拟研究了变形孪晶与纳米结构Cu / Ag界面的相互作用,分析了界面结构对孪晶形核、扩展和增厚的影响,并阐述了失配界面位错网格的作用[4]。图4 显示了肖克利部分位错网格(由黑线突出显示),三角形图案(白色区段)以及界面处的堆垛层错分布。其中,原子根据中心对称参数着色,蓝色的原子处于完美的FCC环境中,而红色的原子处于层错或孪晶断层中。

图5为应力应变曲线和孪晶的原子比例与应变的函数关系。通过分析,他们发现界面可以直接或间接地通过Lomer位错诱导孪晶位错的成核,以及异相界面结构如何影响机械孪晶过程的不同步骤,从而影响纳米结构Cu / Ag中形成的孪晶的大小。通过这种原子尺度方法,为纳米级复合材料中的机械孪晶过程提供了一些有用的理论依据。

设计多层材料来调整机械性能是一个热门话题,同时控制变形机制,因为孪晶允许结合纳米层和纳米孪晶材料的机械性质。在这方面,这项研究提供了理解双界面相互作用机制的关键,并支持了异相界面促进孪晶的观点。
对于包含低对称性hcp 结构金属的超细尺度层状复合材料,由于大量的异质界面能够有效地吸收核辐照引起的空位、间隙原子等缺陷,且hcp金属自身具有密度低、比强度和比刚度高、导电导热性好等特点,近年来由Ti、Zr、Mg与其它金属复合而成的六方系多层材料开始受到人们的关注。但是与晶体结构对称性高的fcc 和bcc 金属相比,hcp金属的室温塑性变形能力差,制约了相关复合材料的使用[1]。
除了原子尺度的空间和时间分辨率,分子动力学模拟一方面可以阐述完全表征的理想化纳米晶体模型的界面结构、驱动力和原子机制等行为;另一方面可以在非常高的晶界和位错密度下观测大塑性变形行为。例如,位错形核机制、晶界湮灭、纳米晶Al中的机械孪晶、减小晶粒尺寸时从位错到基于晶界的变形机制、剪切带的观测及其与凹陷断裂面的关系。
此外,在实际应用和研究过程中,对于不同的问题描述和选用的理论模型,动力学已发展了很多的理论分支,比如,宾夕法尼亚大学的Jian Han, Spencer L. Thomas等人依赖于晶界动力学的断开描述总结了多晶材料的晶界动力学概念[5],Zheng Ma等人对FeCO3进行了析出动力学研究[6],还有表面/界面动力学[7]等。

来源:材料人


